
点击上方“Deephub Imba”,关注公众号,好文章不错过 !
使用散点图评估数值变量之间的相关性很简单,但是分类变量呢?
散点图是很好的可视化工具,用于评估数值或连续变量之间的关系和关联。但是使用数据点来评估分类变量可能并不那么简单。
考虑一个常见的场景,研究人员想要在微阵列(包含约 20,000 个转录本)中找出实验条件 A 是否引发与条件 B 相同的基因表达谱。绘制条形图或箱形图以可视化所有基因表达差异和趋势。但是因存在大量数据点,全部绘制在一张图上会对性能有极大的挑战。
聚类图或热图可能是另一种可视化基因表达差异的替代方法。这些图表没有提供统计数据来衡量基因表达差异的趋势是相似还是不同。所以为了解决这些限制可以使用相关矩阵和pairplot,我们使用Seaborn 可以轻松的绘制他们。如果使用原始值,需要通过计算 log2–transformed fold-change (log2FC) 值来标准化数据。如果数据是带有时间性质,则可以根据基线(时间 = 0)计算 log2FC。
在这篇文章中,我们将使用 Seaborn 和 matplotlib 库:
import csv
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
这里我们将使用Zak等人2012年发表的transcriptomics 数据集,研究血清阳性和血清阴性受试者在不同时间点对Merck Ad5/HIV疫苗的反应。该研究的总结可以在本篇文章的引用中找到,Partek Genomics Suite分析的处理数据集可以在GitHub中找到。这里我们根据基线(时间点= 0)计算fold change, ratio, p-value和adjusted p-value (q-value)。
在这篇文章中,我们将分析Merck Ad5/HIV疫苗接种后不同时间点之间的相关性。
我们将从GitHub加载和处理过的数据文件。将基因列标记为索引列以供参考是很重要的。代码如下:
df = pd.read_csv('https://raw.githubusercontent.com/kuanrongchan/vaccine-studies/main/Ad5_seroneg.csv',index_col=0)
df.head()
输出文件显示了与基线(时间点= 0)相比的6小时、1天、3天和7天时间点的p-value (pval)、调整后的p-value (qval)、比率和折叠变化(fc)的值:
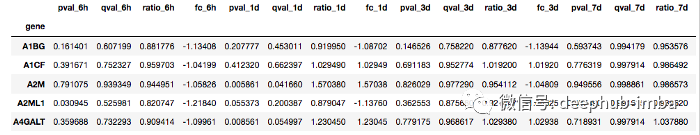
由于log2FC值近似于正态分布或对数正态分布,这些值最适合用于分类变量之间的相关性。因此,我们将把log2FC值制成表格,并过滤包含各个时间点的log2FC值。
df['log2FC_6h'] = np.log2(df['ratio_6h'])
df['log2FC_1d'] = np.log2(df['ratio_1d'])
df['log2FC_3d'] = np.log2(df['ratio_3d'])
df['log2FC_7d'] = np.log2(df['ratio_7d'])df_log2FC = df.filter(items=['log2FC_6h','log2FC_1d', 'log2FC_3d', 'log2FC_7d'])
df_log2FC
输出文件如下:
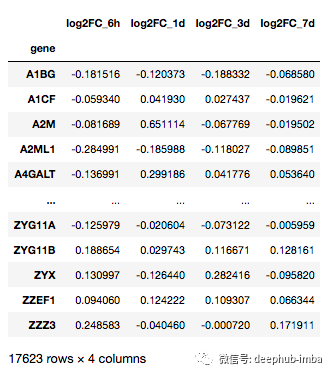
计算不同时间点之间的相关系数,代码如下:
corr = df_log2FC.corr()
corr
显示相关系数的输出为:
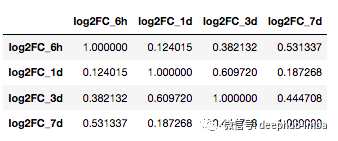
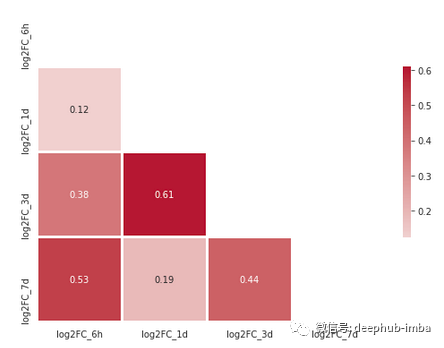
数据表明,第 1 天的基因特征与第 3 天最相似。有趣的是,疫苗接种后 6 小时的特征也与第 7 天相似。
接下来,我们可以评估相关性的p值,以检验相关性的显著性。我们导入SciPy并执行如下代码:
from scipy import stats
from scipy.stats import pearsonr
pvals = pd.DataFrame([[pearsonr(df_log2FC[c], df_log2FC[y])[1] for y in df_log2FC.columns] for c in df_log2FC.columns],
columns=df_log2FC.columns, index=df_log2FC.columns)
pvals
输出是这样的:
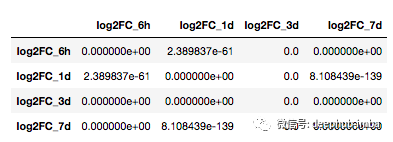
请注意,所有的相关性都是显著的,可能是因为统计分析考虑了大量的数据点。
为了在相关矩阵中显示这些相关系数,我们可以使用以下代码:
mask = np.zeros_like(corr)
mask[np.triu_indices_from(mask)] = True
with sns.axes_style('white'):
f, ax = plt.subplots(figsize=(10, 7))
ax = sns.heatmap(corr, cmap='vlag', mask=mask, center=0, square=True, linewidths=2, annot=True, cbar_kws={'shrink': .5})
简单解释一下上面的代码。这些代码在相关矩阵的上半部分和相同变量之间的相关性上添加一个掩码,以便用户可以专注于图下半部分的比较。我还定义了图形大小、使用的颜色图(范围从蓝色到红色,其中蓝色为负相关,红色为正相关),并将相关值集中在 0(白色)处。annot(注释) 提供图形中的相关系数值,而linewidth (线条宽度) 使我们能够更好地分隔正方形。
相关矩阵的输出如下所示:
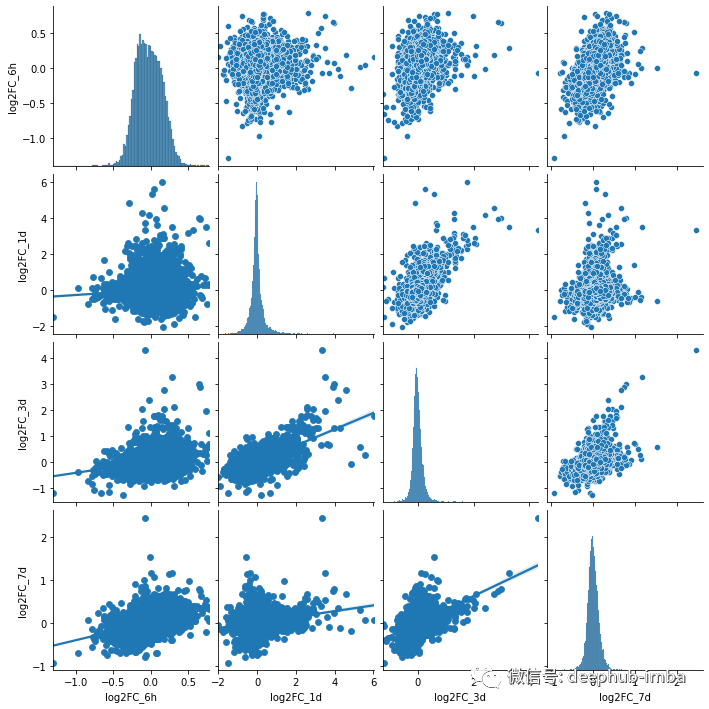
为了让我们看到组成相关矩阵的点,我们可以使用下面的命令来绘制pair plot:
g = sns.pairplot(df_log2FC)
g.map_lower(sns.regplot)
pairplot主要展示变量之间的两两关系,它的下半部分将包含回归图,以便更清楚地可视化趋势和斜率。这在有大量数据点情况下尤为重要。输出文件如下:
最后是引用
https://github.com/kuanrongchan/vaccine-studies/blob/main/Ad5_seroneg.csv
作者:Kuan Rong Chan
喜欢就关注一下吧:
 点个 在看 你最好看!********** **********
点个 在看 你最好看!********** **********