

“脱氧核糖核酸(DNA)是一种分子,其中包含每个物种独特的生物学指令。DNA及其包含的说明在繁殖过程中从成年生物传给其后代。“ —genome.gov
简介
基因组是生物体中DNA的完整集合。所有生物物种都有一个基因组,但是它们的差异很大。例如,人类基因组被排列成23条染色体,这有点像百科全书被编辑成23卷。如果算上所有字符(单个DNA“碱基对”),每个人类基因组中将有超过60亿个字符。所以这是一个巨大的工程。
人类基因组大约有60亿个字符。如果您认为基因组(完整的DNA序列)就像一本书,那就是一本由大约60亿个“ A”,“ C”,“ G”和“ T”字母组成的书。每个人都有独特的基因组。尽管如此,科学家发现人类基因组的大部分彼此相似。
作为数据驱动的科学,基因组学广泛地利用机器学习来捕获数据中的关系并推断出新的生物学假设。但是,要想拥有从不断增长的基因组学数据中提取新发现的能力,就需要更强大的机器学习模型。通过有效利用大型数据集,深度学习已给了计算机视觉和自然语言处理等领域带来了很大进步。它已成为许多基因组建模任务的首选方法,包括预测遗传变异对基因调控机制(如DNA接受性和DNA剪接)的影响。
在本文中,我们将了解如何解释DNA结构以及如何使用机器学习算法来建立DNA序列数据的预测模型。
DNA序列如何表示?
该图显示了DNA双螺旋结构的一小部分。
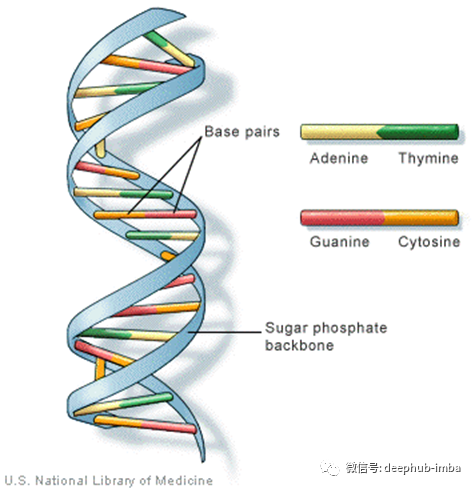
DNA的双螺旋结构
双螺旋是DNA的化学表示。但是DNA很特别。它是由四种类型的碱基组成的核苷酸:**腺嘌呤(A),胸腺嘧啶(T),鸟嘌呤(G)和胞嘧啶(C)**。我们总是称它们为
A
,
C
,
G
和
T
。
这四种化学物质通过氢键以任何可能的顺序连接在一起,形成一条链,这形成了DNA双螺旋的一条线。双螺旋的第二条线平衡了第一根。因此,如果第一条线上有A,则第二条线上对应位置必须为T。此外,C和G始终保持平衡。因此,一旦您确定了螺旋的一个螺纹,就可以随时拼写另一个螺纹。

2
单个DNA线(螺旋)的序列
这些碱基的顺序决定了DNA链中包含哪些生物学指令。例如,序列ATCGTT可能指示蓝眼睛,而ATCGCT可能指示棕色眼睛。
使用Python处理DNA序列数据
熟悉诸如
Biopython
和
squiggle
之类的Python包将在处理Python中的生物序列数据时为您提供帮助。
Biopython是python模块的集合,这些模块提供处理DNA,RNA和蛋白质序列操作的功能,例如DNA字符串的反向互补,寻找蛋白质序列中的基序列等。它提供了很多解析器,可以读取所有主要的遗传数据库,例如 GenBank,SwissPort,FASTA等,

4
安装Biopython
pip install biopython
**Squiggle:**这是一个工具,可以自动生成交互式网页下的原始DNA序列的二维图形表示。考虑到易用性,Squiggle实现了几种序列可视化算法,并引入了“为方便人类使用而做”的新颖可视化方法。
安装Squiggle
pip install Squiggle
DNA序列数据通常以“ fasta”格式的文件格式储存。Fasta格式通过包含注释的大于号和包含序列的另一行作为组成自己的单行数据:
“AAGGTGAGTGAAATCTCAACACGAGTATGGTTCTGAGAGTAGCTCTGTAACTCTGAGG”*
文件可以包含一个或多个DNA序列。还有许多其他格式,但是fasta是最常见的格式。
这是使用Biopython处理Fasta格式的DNA序列的简要示例。序列对象将包含诸如序列ID和sequence等属性以及可以直接使用的序列长度。
我们将使用Biopython的
Bio.SeqIO
来解析DNA序列数据(fasta)。它提供了一个简单的统一界面来输入和输出各种文件格式。
from Bio import SeqIOfor for sequence in SeqIO.parse('./drive/My Drive/example.fa', "fasta"): print(sequence.id) print(sequence.seq) print(len(sequence))
这样就产生了序列ID,序列本身和序列长度。
ENST00000435737.5ATGTTTCGCATCACCAACATTGAGTTTCTTCCCGAATACCGACAAAAGGAGTCCAGGGAATTTCTTTCAGTGTCACGGACTGTGCAGCAAGTGATAAACCTGGTTTATACAACATCTGCCTTCTCCAAATTTTATGAGCAGTCTGTTGTTGCAGATGTCAGCAACAACAAAGGCGGCCTCCTTGTCCACTTTTGGATTGTTTTTGTCATGCCACGTGCCAAAGGCCACATCTTCTGTGAAGACTGTGTTGCCGCCATCTTGAAGGACTCCATCCAGACAAGCATCATAAACCGGACCTCTGTGGGGAGCTTGCAGGGACTGGCTGTGGACATGGACTCTGTGGTACTAAATGAAGTCCTGGGGCTGACTCTCATTGTCTGGATTGACTGA390
我们可以可视化这些DNA序列吗?
是的,我们可以使用Squiggle python库将这些DNA序列可视化,进行操作并运行:
Squiggle example.fa --method=gates
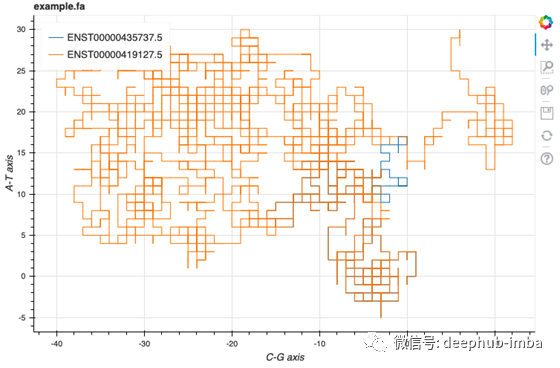
DNA序列被转换为2D图像,其中T,A,C和G分别在上,下,左和右方位。这给每个序列一个“形状”。
现在,我们来可视化另一个包含6个DNA序列的fasta数据。
Squiggle example.fasta
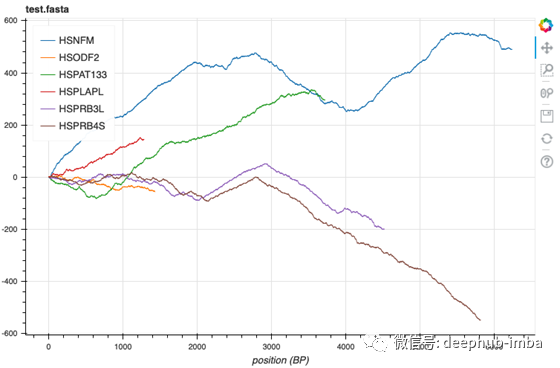
在此,首先使用2位编码方案将DNA序列转换为二进制序列,该方案将T映射为00,C映射为01,A映射为10,G映射为11。
现在我们可以轻松加载和操作生物序列数据,那么**怎么将数据用于机器学习或深度学习**?
由于机器学习或深度学习模型要求输入必须是特征矩阵或数字值,但目前我们仍然以字符或字符串格式存储数据。因此,下一步是将这些字符编码为矩阵。
编码序列数据有3种通用方法:
顺序编码DNA序列
独热(one-hot)编码DNA序列
DNA序列作为独立“语言”,称为k-mer计数
让我们分别进行实现,看看哪一个为我们提供了完美的输入。
顺序编码DNA序列
在这种方法中,我们需要将每个碱基编码为序数值。例如,“ ATGC”变为[0.25、0.5、0.75、1.0]。任何其他字符(例如“ N”)都可以为0。
因此,让我们创建一些函数,例如从序列字符串创建NumPy数组对象,以及带有DNA序列字母“ a”,“ c”,“ g”和“ t”的标签编码器,以及其他任何字符比如“n”的编码器。
import numpy as npimport redef string_to_array(seq_string): seq_string = seq_string.lower() seq_string = re.sub('[^acgt]', 'n', seq_string) seq_string = np.array(list(seq_string)) return seq_string# create a label encoder with 'acgtn' alphabetfrom sklearn.preprocessing import LabelEncoderlabel_encoder = LabelEncoder()label_encoder.fit(np.array(['a','c','g','t','z']))
这是一种将DNA序列字符串编码为有序载体的功能。它返回一个NumPy数组,其中A = 0.25,C = 0.50,G = 0.75,T = 1.00,n = 0.00。
def ordinal_encoder(my_array): integer_encoded = label_encoder.transform(my_array) float_encoded = integer_encoded.astype(float) float_encoded[float_encoded == 0] = 0.25 # A float_encoded[float_encoded == 1] = 0.50 # C float_encoded[float_encoded == 2] = 0.75 # G float_encoded[float_encoded == 3] = 1.00 # T float_encoded[float_encoded == 4] = 0.00 # anything else, lets say n return float_encoded
让我们尝试一个简单的序列:
seq_test = 'TTCAGCCAGTG'ordinal_encoder(string_to_array(seq_test))

独热编码DNA序列
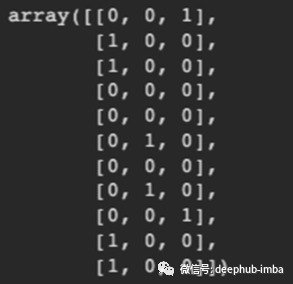
另一种方法是使用独热编码来表示DNA序列。这在深度学习方法中得到了广泛使用,非常适合卷积神经网络之类的算法。在此示例中,“ ATGC”将变为[0,0,0,1],[0,0,1,0],[0,1,0,0],[1,0,0,0]。这些编码的矢量可以连接起来,也可以变成二维数组。
from sklearn.preprocessing import OneHotEncoderdef one_hot_encoder(seq_string): int_encoded = label_encoder.transform(seq_string) onehot_encoder = OneHotEncoder(sparse=False, dtype=int) int_encoded = int_encoded.reshape(len(int_encoded), 1) onehot_encoded = onehot_encoder.fit_transform(int_encoded) onehot_encoded = np.delete(onehot_encoded, -1, 1) return onehot_encoded
让我们以一个简单的序列来尝试一下:
seq_test = 'GAATTCTCGAA'one_hot_encoder(string_to_array(seq_test))
DNA序列作为独立“语言”,称为k-mer计数
仍然存在的问题是,以上所有方法均不能产生长度一致的向量,这是将数据导入分类或回归算法的必要条件。因此,使用上述方法,您必须辅助诸如截断序列或用“ n”/“ 0”填充的方法,以获取长度一致的向量。
DNA和蛋白质序列可以看作是生命的语言。该语言对所有生命形式中存在的分子的指令和功能进行编码。基因组与序列语言和书是相似的,子序列(基因和基因家族)是句子和章节,k-mers和肽是单词,核苷酸碱基和氨基酸是字母。自然语言处理(NLP)也应采用和DNA及蛋白质序列相似的处理方式是有理由的。
我们在这里使用的方法是易于管理的。我们首先采用较长的生物学序列,并将其分解为k-mer长度重叠的“单词”。例如,如果我们使用长度为6(六进制)的“单词”,则“ ATGCATGCA”将变为:“ ATGCAT”,“ TGCATG”,“ GCATGC”,“ CATGCA”。因此,我们的示例序列分为4个六聚体字(hexamer words)。
在基因组学中,我们将这种类型的操作称为“ k-mer计数”,或者对每种可能出现的k-mer序列进行计数,而Python的自然语言处理工具使其变得非常容易。
def Kmers_funct(seq, size): return [seq[x:x+size].lower() for x in range(len(seq) - size + 1)]
因此,让我们以一个简单的序列尝试一下:
mySeq = 'GTGCCCAGGTTCAGTGAGTGACACAGGCAG'Kmers_funct(mySeq, size=7)

它返回k-mer“单词”的列表。然后,您可以将“单词”加入到“句子”中,然后像往常一样在“句子”上应用自己喜欢的自然语言处理方法。
words = Kmers_funct(mySeq, size=6)joined_sentence = ' '.join(words)joined_sentence
连接的句子:
'gtgccc tgccca gcccag cccagg ccaggt caggtt aggttc ggttca gttcag ttcagt tcagtg cagtga agtgag gtgagt tgagtg gagtga agtgac gtgaca tgacac gacaca acacag cacagg acaggc caggca aggcag'
您可以调整单词长度和重叠量。这使您可以确定DNA序列信息和词汇量在您的应用程序中的重要程度。例如,如果您使用长度为6的单词,并且有4个字母,则词汇量为4096个可能的单词。然后,您可以像在NLP中一样继续创建单词库(bag-of-words)模型。
让我们增加一些更有趣的“句子”。
mySeq1 = 'TCTCACACATGTGCCAATCACTGTCACCC'mySeq2 = 'GTGCCCAGGTTCAGTGAGTGACACAGGCAG'sentence1 = ' '.join(Kmers_funct(mySeq1, size=6))sentence2 = ' '.join(Kmers_funct(mySeq2, size=6))
建立单词袋模型:
from sklearn.feature_extraction.text import CountVectorizercv = CountVectorizer()X = cv.fit_transform([joined_sentence, sentence1, sentence2]).toarray()X
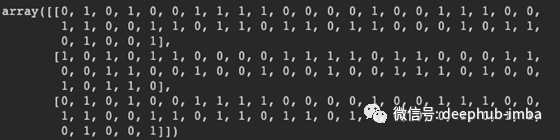
单词袋
下面开始进行机器学习
现在,我们已经学习了如何从DNA序列中提取特征矩阵,让我们将我们新获得的知识应用于机器学习用例。

用例:建立一个在人类DNA序列上受训的分类模型,并可以根据编码序列的DNA序列预测基因家族。为了测试该模型,我们将使用人,狗和黑猩猩的DNA序列进行训练,并测试其准确性。
基因家族是一组具有共同祖先的相关基因。基因家族的成员可以是旁系同源物或直系同源物。基因旁系同源物是来自相同物种的具有相似序列的基因,而基因直系同源物是在不同物种中具有相似序列的基因。
数据集包含人类的DNA序列,狗的DNA序列和黑猩猩的DNA序列。
加载人类DNA序列。
import numpy as npimport pandas as pdimport matplotlib.pyplot as plt%matplotlib inlinehuman_dna = pd.read_table('./drive/My Drive/human_data.txt')human_dna.head()

人类DNA序列和类别标签
加载黑猩猩和狗的DNA序列。
chimp_dna = pd.read_table('./drive/My Drive/chimp_data.txt')dog_dna = pd.read_table('./drive/My Drive/dog_data.txt')

狗DNA序列和类别标签
这是7个类别中每个类别的定义,以及人工训练数据中的类别数:

人类DNA数据集中存在带有类别标签的基因家族
现在我们已经加载了所有数据,下一步是将字符序列转换为k-mer词,默认大小为6(六进制)。函数
Kmers_funct()
将从序列字符串中收集指定长度的所有可能重叠的k-mers。
def Kmers_funct(seq, size=6): return [seq[x:x+size].lower() for x in range(len(seq) - size + 1)]#convert our training data sequences into short overlapping k-mers of length 6. Lets do that for each species of data we have using our Kmers_funct function.human_dna['words'] = human_dna.apply(lambda x: Kmers_funct(x['sequence']), axis=1)human_dna = human_dna.drop('sequence', axis=1)chimp_dna['words'] = chimp_dna.apply(lambda x: Kmers_funct(x['sequence']), axis=1)chimp_dna = chimp_dna.drop('sequence', axis=1)dog_dna['words'] = dog_dna.apply(lambda x: Kmers_funct(x['sequence']), axis=1)dog_dna = dog_dna.drop('sequence', axis=1)
将DNA序列更改为小写,分为所有可能的长度为6的k-mer字,并准备下一步。
human_dna.head()

人类DNA序列中长度为6的k-mer字
现在,我们需要将每个基因的k-mers列表转换为可用于创建单词袋模型的字符串句子。我们将创建一个目标变量
y
来保存类标签。
对黑猩猩和狗也进行一样的操作。
human_texts = list(human_dna['words'])for item in range(len(human_texts)): human_texts[item] = ' '.join(human_texts[item])#separate labelsy_human = human_dna.iloc[:, 0].values # y_human for human_dna#Now let's do the same for chimp and dog.chimp_texts = list(chimp_dna['words'])for item in range(len(chimp_texts)): chimp_texts[item] = ' '.join(chimp_texts[item])#separate labelsy_chim = chimp_dna.iloc[:, 0].values # y_chim for chimp_dnadog_texts = list(dog_dna['words'])for item in range(len(dog_texts)): dog_texts[item] = ' '.join(dog_texts[item])#separate labelsy_dog = dog_dna.iloc[:, 0].values # y_dog for dog_dna
因此,目标变量包含一个保存类别的数组。
array([4, 4, 3, …, 6, 6, 6])
将我们的k-mer单词转换为均等长度的数字矢量,这些矢量代表词汇中每个k-mer的计数:
from sklearn.feature_extraction.text import CountVectorizercv = CountVectorizer(ngram_range=(4,4)) #The n-gram size of 4 is previously determined by testingX = cv.fit_transform(human_texts)X_chimp = cv.transform(chimp_texts)X_dog = cv.transform(dog_texts)
您可能要检查每个训练集数据的形状。
print(X.shape)print(X_chimp.shape)print(X_dog.shape)#shapes(4380, 232414) (1682, 232414) (820, 232414)
因此,对于人类,我们已经将4380个基因转换为4-gram的k-mer(长度6)计数的均匀长度特征向量。对于黑猩猩和狗,我们分别具有1682和820个基因的相同形状的特征。
既然我们知道如何将我们的DNA序列转换为k-mer计数和n-gram形式的均匀长度的数字矢量,那么我们现在就可以继续构建一个分类模型,该模型可以仅基于序列本身来预测DNA序列功能 。
在这里,我将使用人类数据来训练模型,并拿出20%的人类数据来测试模型。然后,通过尝试预测其他物种(黑猩猩和狗)的序列功能,我们可以测试模型的可推广性。
接下来,拆分用来训练/测试的人类数据集并构建简单的多项朴素贝叶斯分类器。
您可能需要进行一些参数调整,并构建具有不同n-gram大小的模型,在这里,我将继续使用n-gram大小为4和alpha为0.1的模型。
# Splitting the human dataset into the training set and test setfrom sklearn.model_selection import train_test_splitX_train, X_test, y_train, y_test = train_test_split(X, y_human, test_size = 0.20, random_state=42)## Multinomial Naive Bayes Classifier ##from sklearn.naive_bayes import MultinomialNBclassifier = MultinomialNB(alpha=0.1)classifier.fit(X_train, y_train)
现在,让我们对测试集进行预测,看看它的性能如何。
y_pred = classifier.predict(X_test)
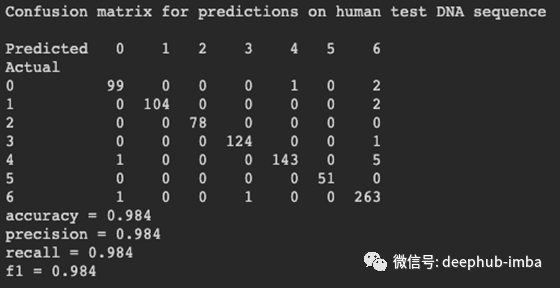
下面让我们来看一些模型性能指标,例如混淆矩阵,准确性,召回率和f1得分。我们在数据上获得了非常好的结果,因此看来我们的模型并未过拟合训练数据。
from sklearn.metrics import accuracy_score, f1_score, precision_score, recall_scoreprint("Confusion matrix for predictions on human test DNA sequence\n")print(pd.crosstab(pd.Series(y_test, name='Actual'), pd.Series(y_pred, name='Predicted')))def get_metrics(y_test, y_predicted): accuracy = accuracy_score(y_test, y_predicted) precision = precision_score(y_test, y_predicted, average='weighted') recall = recall_score(y_test, y_predicted, average='weighted') f1 = f1_score(y_test, y_predicted, average='weighted') return accuracy, precision, recall, f1accuracy, precision, recall, f1 = get_metrics(y_test, y_pred)print("accuracy = %.3f \nprecision = %.3f \nrecall = %.3f \nf1 = %.3f" % (accuracy, precision, recall, f1))
让我们看看我们的模型如何处理其他物种的DNA序列。首先,我们将尝试黑猩猩,我们希望它与人类非常相似。然后是狗DNA序列。
# Predicting the chimp, dog and worm sequencesy_pred_chimp = classifier.predict(X_chimp)
检查精度矩阵:
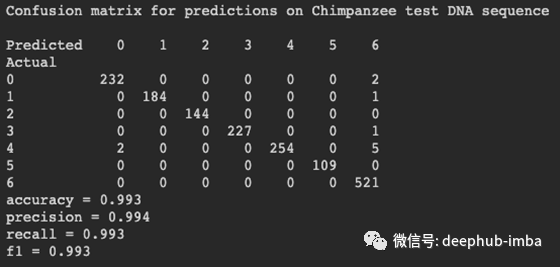
# performance on chimpanzee genesprint("Confusion matrix for predictions on Chimpanzee test DNA sequence\n")print(pd.crosstab(pd.Series(y_chim, name='Actual'), pd.Series(y_pred_chimp, name='Predicted')))accuracy, precision, recall, f1 = get_metrics(y_chim, y_pred_chimp)print("accuracy = %.3f \nprecision = %.3f \nrecall = %.3f \nf1 = %.3f" % (accuracy, precision, recall, f1))
现在让我们对狗的测试DNA序列进行预测:
y_pred_dog = classifier.predict(X_dog)
检查精度矩阵:
# performance on dog genesprint("Confusion matrix for predictions on Dog test DNA sequence\n")print(pd.crosstab(pd.Series(y_dog, name='Actual'), pd.Series(y_pred_dog, name='Predicted')))accuracy, precision, recall, f1 = get_metrics(y_dog, y_pred_dog)print("accuracy = %.3f \nprecision = %.3f \nrecall = %.3f \nf1 = %.3f" % (accuracy, precision, recall, f1))
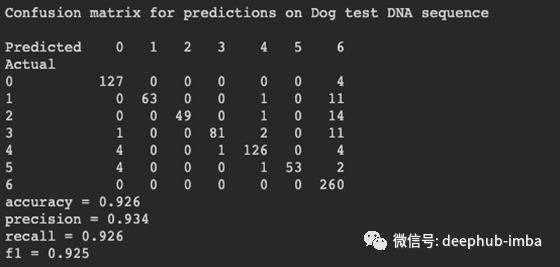
该模型似乎在人类数据上产生了良好的结果。黑猩猩也是如此,这是因为黑猩猩和人类具有相同的遗传层次。狗的表现不太好,这是因为狗比黑猩猩与人类的差异更大。
结论
在本文中,我们学习了如何分析DNA序列数据,如何对其进行可视化,以及如何使用不同的编码技术将这些序列表示为矩阵。最后,我们创建了一个Naive Byes模型,可以在人,狗和黑猩猩的测试数据中检测基因家族。
免责声明
本文只是从机器学习的角度探讨DNA的测序辅助,本人也并非医学的专业人士,所以涉及医学的部分还应以医学专业人员为准。
本文github代码地址:https://github.com/nageshsinghc4/DNA-Sequence-Machine-learning
DeepHub
微信号 : deephub-imba
每日大数据和人工智能的重磅干货
大厂职位内推信息
长按识别二维码关注 ->
好看就点在看!********** **********