
脑部疾病的异质性是精准诊断/预后的一个挑战。作者描述并验证了一种名为Smile-GAN(SeMI-supervised cLustEring-Generative Adversarial Network),的半监督深度聚类方法,它研究了与正常大脑结构对比的神经解剖学异质性,从而通过神经影像特征识别疾病亚型。当应用于来自T1加权MRI的区域体积时(两项研究;2832名参与者;8146次扫描),包括认知正常的人和那些有认知障碍和痴呆症的人,Smile-GAN确定了四种神经变性模式。将此框架应用于纵向数据揭示了两种不同的进展途径。这些模式的表达测量预测了未来神经变性的途径和速度。模式表达在预测临床进展方面提供了与淀粉样/tau蛋白互补的性能。这些深度学习衍生的生物标志物为精确诊断和有针对性的临床试验招募提供了潜力。
介绍
神经系统和神经精神系统疾病与障碍的神经影像学和临床表型通常非常异质。人工智能方法,尤其是深度学习方法,在医学成像应用中取得了显着的飞跃,并在获得具有诊断和预后价值的解剖学、功能和病理学的个性化神经成像特征方面显示出巨大的前景。然而,直到最近才开发出深度学习方法,通过识别可能具有不同预后、进展模式和对治疗反应的常见但不同的疾病亚型来研究疾病的异质性。为了实现这一目标,此研究提出了一种半监督的深度学习范式(图1),称为Smile-GAN(通过生成对抗网络的半监督聚类)。Smile-GAN 通过对正常测量的稀疏变换来模拟疾病影响,利用经过训练的 GAN 来合成变换,从而产生难以与来自真实患者数据的测量区分开来的真实测量。估计的潜在变量捕获表型亚型,以反向一致的公式调节这种合成,确保可以从各自的生物标志物特征可靠地估计亚型成员。
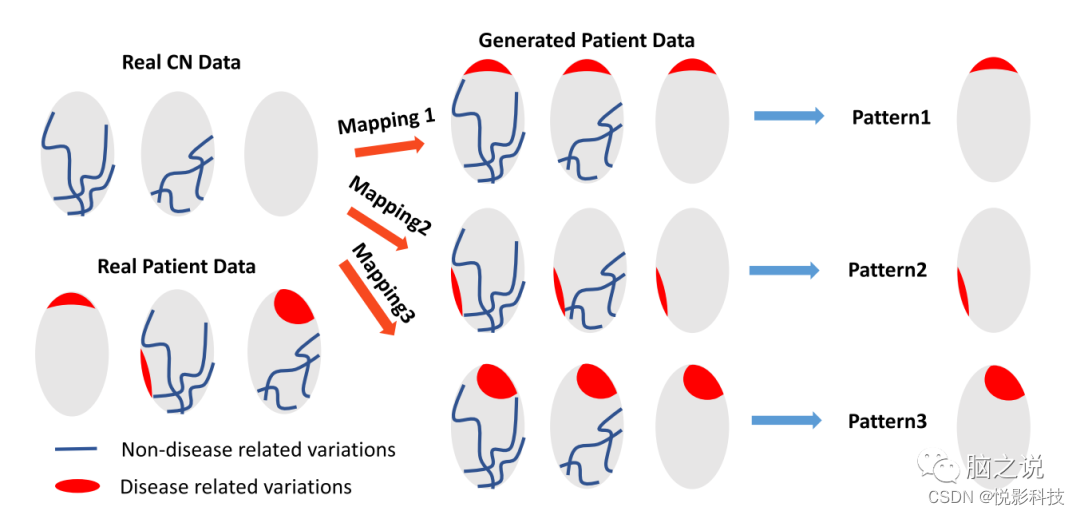
图1. Smile-GAN 的概念概述。蓝线代表在正常对照组 (CN) 和患者组中观察到的非疾病相关变化。红色区域代表仅存在于患者组中的疾病影响。Smile-GAN 通过从 CN 数据到患者数据的聚类转换来发现神经解剖模式类型。
虽然Smile-GAN是一种通用方法,但在此测试它识别大脑神经解剖学中的异质性——特别是通过灰质和白质感兴趣区域体积的减少和心室体积的增加来衡量的萎缩的异质性——在2个纵向队列(ADNI和BLSA)的2832名个体的 8146次扫描中的从早期认知障碍到痴呆的整个光谱中发现的。阿尔茨海默病 (AD)是最常见的神经变性疾病,影响全球数百万人,并占此研究样本中认知能力下降的大部分。AD的标志性病理学包括存在ß-淀粉样神经炎斑块和含tau蛋白的神经原纤维缠结,这有助于在磁共振成像(MRI)测量的特征性神经变性。虽然诊断标准传统上侧重于临床综合征,通常主要是AD的遗忘表型和称为轻度认知障碍(MCI)的痴呆前阶段,但最近人们越来越努力基于淀粉样蛋白沉积(A)、tau沉积(T)和神经变性(N)的生物标志物的存在从生物学上定义AD,每个标志物的特征通常是二分法,即不存在(-)或存在(+),从而定义了AT(N)框架。虽然有用,但这种二元表征很难捕捉生物标志物的异质性,例如AD解剖图的已知变异性或常见共病的影响,包括血管疾病和其他可能以不同方式影响“N”维度的共病神经退行性过程。这种可变性以及患者对神经病理学的适应能力在个体认知衰退的最终表现中起着重要作用,因此对于理解从疾病的群体影响转向个性化诊断时至关重要。此外,通过更清楚地识别神经变性的典型模式和严重程度,包括更多暗示潜在AD的模式,这种方法可以改进临床试验参与者的选择。
几种MRI生物标志物已被用于量化AD中的神经变性。最常见的一种是海马体积;海马萎缩是典型AD的特征。然而,对于其他单个感兴趣区域 (ROI) 标记,它既不是AD特有的,也不是捕获与整体表型相关的复杂的大脑萎缩模式。对AD中常见的颞顶萎缩敏感的综合测量,包括各种区域体积特征或SPARE-AD 等机器学习指标,提供神经变性的替代措施,这些措施还可以捕捉多个大脑区域的相关变化。这些方法提供了具有高度敏感性和特异性的类似AD的神经变性的整体特征,但没有阐明AD及其临床前阶段中发现的神经变性模式的异质性,也没有试图将这种异质性与共病病理联系起来。最近,利用大型神经影像数据集和新型机器学习方法的新型数据驱动方法已经出现,以识别AD和其他神经变性疾病中的脑萎缩模式。
聚类方法已用于识别患者的横截面或时间异质性。Zhang等人使用贝叶斯潜在狄利克雷分配(LDA)模型从源自结构MRI的体素灰质(GM)密度图中识别潜在萎缩模式。Young等人提出通过推断亚型和阶段来揭示时间和表型异质性。然而,这些方法仅基于患者得出聚类,因此可以识别部分包含影响个体间大脑变异的与疾病无关的混杂因素的聚类或模式。此研究提出的半监督方法旨在通过有效地聚集认知正常(CN)个体和患者之间的差异来克服这一限制,从而专注于病理过程的神经解剖学异质性,而不是可能由各种混杂因素引起的异质性。生成对抗网络(GAN)以使用两个神经网络之间的竞争来学习和建模复杂分布而闻名。该研究中,作者使用GAN 来合成源自成像数据的异常真实的区域体积,以模拟疾病影响并执行半监督聚类。
建立在基于GAN的模型,Smile-GAN方法通过转换CN个体的基于ROI的神经解剖数据来生成真实的ROI体积数据,从而捕获不同的疾病相关的神经解剖模式。通过反向一致的潜在变量,这种合成由与疾病相关的神经解剖亚型引导的,这些亚型是根据数据估计出来的。此外,作者使用模拟数据以及脑萎缩的合成模式广泛验证了这种方法。Smile-GAN不直接生成疾病阶段的预测,而是量化捕获模式的表达程度。混合病理和分期可以通过衍生模式或其组合的表达的事后、第二阶段分析来捕获。
作者假设Smile-GAN在对富含AD的样本进行训练时,会识别出患者在AD通路中常见的神经变性模式。作者在认知衰退的范围内发现了4种可重现的神经解剖学萎缩模式,并开发了量化任何个体中每种模式的表达水平的方法,从而形成了一个4维系统,可捕捉到AT(N)系统中“N”维的主要异质性模式。此外,通过测量该坐标系内的纵向轨迹,作者确定了两种不同的进展途径,这意味着存在共病症的存在和/或AD病理过程的异质性的可变性。作者确定了对个体参与者未来神经退行性和临床轨迹具有预测能力的基线模式。
结果
在合成和半合成数据集上验证Smile-GAN模型
在合成数据集上进行的实验验证了该模型能够捕获异质的疾病相关变异,同时不会被非疾病相关变异混淆。映射函数捕获了沿每个方向模拟萎缩的所有区域,同时几乎完美地避开了模拟非疾病相关变化更强的所有区域。半合成数据集的实验,源自真实的MRI ROI数据,但在选定的ROI 中具有人工脑萎缩,进一步验证了模型在更真实的场景下避免非疾病相关变异的能力。此外,该模型的性能被证明优于其他先进的半监督聚类方法和传统的聚类方法,即使在萎缩率非常小和不稳定的情况下也能检测到模拟的模式类型。
神经退行性变的四种模式
根据ADNI2/GO参与者的基线数据进行训练并通过排列测试进行验证,Smile-GAN在认知障碍参与者中确定了四种显着可重复且与疾病相关的脑萎缩模式。使用留出交叉验证实验发现四种模式类型是可重现的。图2b在菱形图中绘制每个参与者属于每个模式的估计概率,每个参与者根据主要模式着色(菱形图足以可视化这4个模式,因为模式概率总和为1,并且没有参与者同时具有这两种模式P1和P4概率>0)。与CN相比,具有不同模式的参与者显示出明显的萎缩特征,这通过CN组与基于概率最高的模式隔离的参与者组之间基于体素的组比较结果显示(图2a)。从这些结果中,可以直观地将四种成像模式解释为:(i)P1,保留的脑容量,与CN 相比在整个大脑中没有表现出明显的萎缩;(ii) P2,轻度弥漫性萎缩,伴有广泛的轻度皮质萎缩,没有明显的内侧颞叶萎缩;(iii) P3,局部内侧颞叶萎缩,显示海马和前内侧颞叶皮质局部萎缩,其他部位相对较少;(iv) P4,晚期萎缩,显示整个大脑严重萎缩,包括严重的颞叶萎缩。当作者对来自各种独立AD研究的参与者进行模型训练时,这四种模式具有高度可重复性,进一步证明这些是AD研究中的保守模式。此外,当作者仅使用具有阳性ß-淀粉样蛋白(Abeta)状态的参与者训练模型时,它们被重现,表明这些四种模式的概率也捕捉到了在有证据表明与AD相关的神经病理变化的参与者中观察到的共同变化。这些模式将可能表现出病理学上确定的AD亚型的参与者分开,由成像生物标志物决定。P3模式包括那些可能患有边缘为主神经病理学的人,P2 包括那些可能患有海马保留神经病理学的人,而混合P2-P3可能是更典型的AD。
两条进展路径
图2c揭示了具有纵向数据的研究子样本中模式概率随时间的演变。在基线具有显着P1特征的参与者可能会在短期内表达P2或P3的概率增加,然后是P4 模式的稍后表达。在基线具有显性P2或P3表达的参与者显示出其他模式的可变次要表达(其他P3/P2模式概率范围从0到0.5)。P2和P3参与者在以后的时间点都有增加的P4概率,但分别没有发展出其他P3或P2模式的显着表达。最初具有最高P4概率的参与者只会随着时间的推移表现出更强的P4表达。从这些结果中,作者得出结论,P1-2-4和P1-3-4是神经变性的两种常规MRI进展途径。图2d在模式维度系统中显示一些代表性参与者随时间推移的详细进展路径。这些例子表明,尽管遵循类似的进展路径,参与者可能在模式纯度和进展速度方面存在差异。更具体地说,虽然用紫色表示的参与者在进展过程中都表现出比P2 更高的P3概率,但实线更接近P2三角形,表明该参与者具有相对更强的P2表达。此外,用虚线表示的参与者在5年内从P1进步到P4(图中未显示时间),而用实线表示的参与者从P1进步到P4需要10年以上。
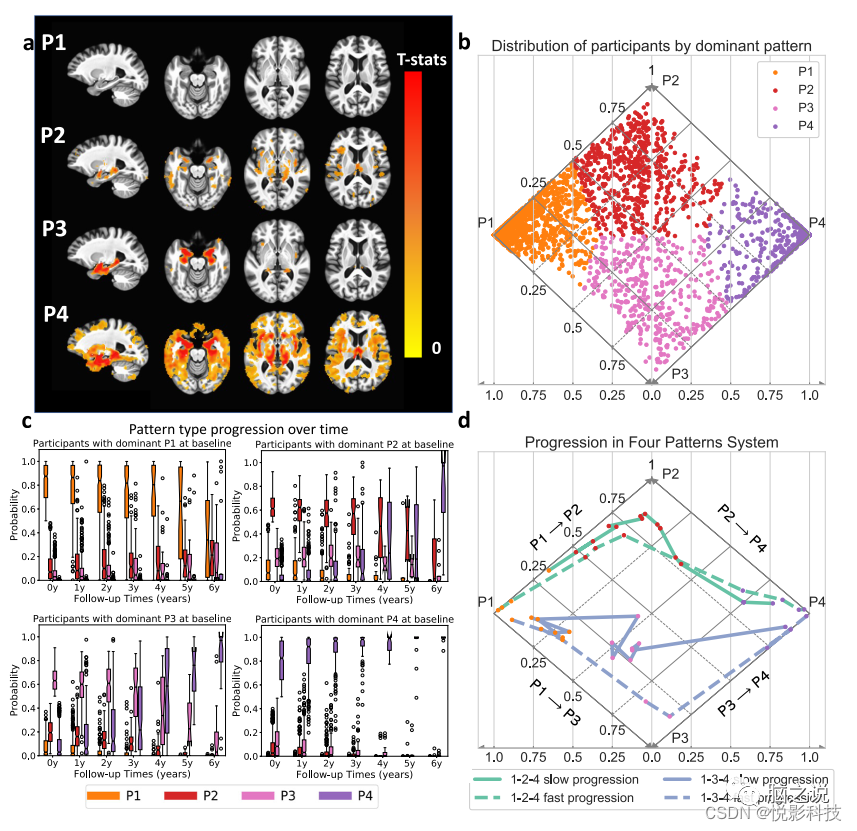
图 2. 四种萎缩模式(P1-P4)和两种神经退行性病变进展途径的表征。(来自发现集(a)中899 名ADNI2/GO参与者和所有2832名ADNI/BLSA参与者(b–d)的数据)。CN和主要属于四种模式中的每一种模式的参与者之间的体素统计比较(单边t检验)。应用了p值阈值为0.05的多重比较的错误发现率(FDR)校正。b参与者在菱形图中表达四种模式的可视化。属于每个模式的伪概率反映了各个模式的表达水平(即存在)和概率亚型成员资格。水平轴表示P1 和P4 概率,对角轴表示P2(实线)和P3(虚线)概率。由于参与者从来没有P1和P4 > 0,所有观察到的模式组合都可以在这个菱形图中表示。个别参与者的点由主要模式进行颜色编码。c每个基线模式组的四种模式随时间的表达盒须图。(中心线,中位数;框限,上下四分位数;胡须,1.5 倍四分位数范围;点,异常值)。d四名代表性参与者的进步路径。虚线表示参与者在5年内从P1达到P4,实线表示那些需要10年以上才能从P1达到P4的参与者。
淀粉样蛋白/tau/模式/诊断
大多数CN参与者的Abeta状态为阴性(A-)并表达为P1(图 3a))。P1还
包括最大数量的认知障碍但非痴呆的参与者,在BLSA/ADNI中分类为MCI,与其他三种模式相比,淀粉样蛋白阴性状态不成比例。患有P2和P3的MCI/痴呆症参与者的数量相当(144和178),他们的淀粉样蛋白状态分布相似,主要是淀粉样蛋白阳性(66.9%和72.1%)。P4参与者大多是淀粉样蛋白阳性(84.0%),很少是CN (3.3%)。模式成员可用于根据 AT(N) 标准对参与者进行分类,从而深入了解疾病的阶段、复原力和共病的存在。沿着AD连续体放置的那些进一步细分为早期神经变性(P3)、晚期(P4)神经变性、或P2组轻度弥漫性萎缩,可通过其他量化方法分类为N-或N+。如图3b所示,根据模式和Abeta/磷酸化-tau (pTau) 状态,参与者被归类为正常,属于典型的AD连续体,属于AD的主要共病学或怀疑非AD病理学(SNAP)。正如预期的那样,A+T+参与者的神经变性往往比 A+T-参与者更严重。使用模式成员作为(N)的分类会适度增加类别的数量(从8 到16),但提供了重要的严重程度和预后信息。
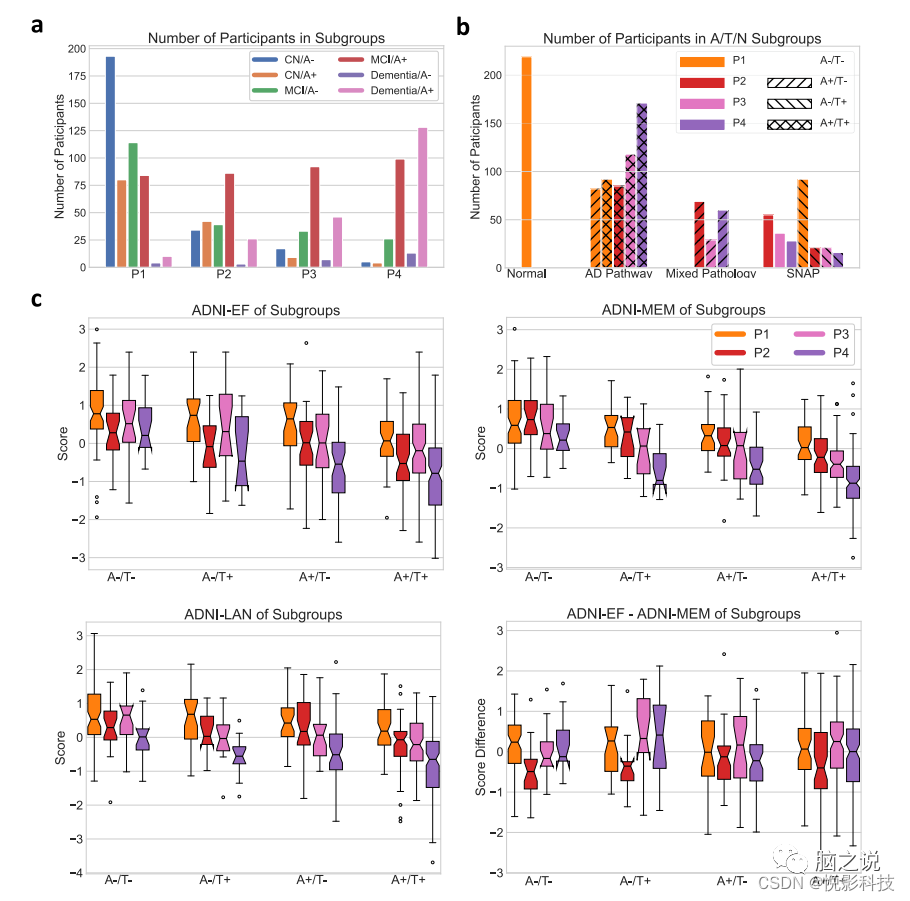
图 3. 参与者分组和子组的认知表现。(来自1194名Abeta/pTau测量的ADNI参与者的数据)a按诊断、淀粉样蛋白状态和模式分组的参与者数量。b基于参与者模式和CSF Abeta/pTau 状态的AT(N)分类。根据模式,N分为正常(P1)、非典型AD (P2)或AD特征(P3/P4)。c按模式划分的MCI/痴呆症参与者认知表现的箱线图。(A:Abeta;T:pTau)(中心线,中位数;框限,上下四分位数;胡须,1.5×四分位数范围;点,异常值)。
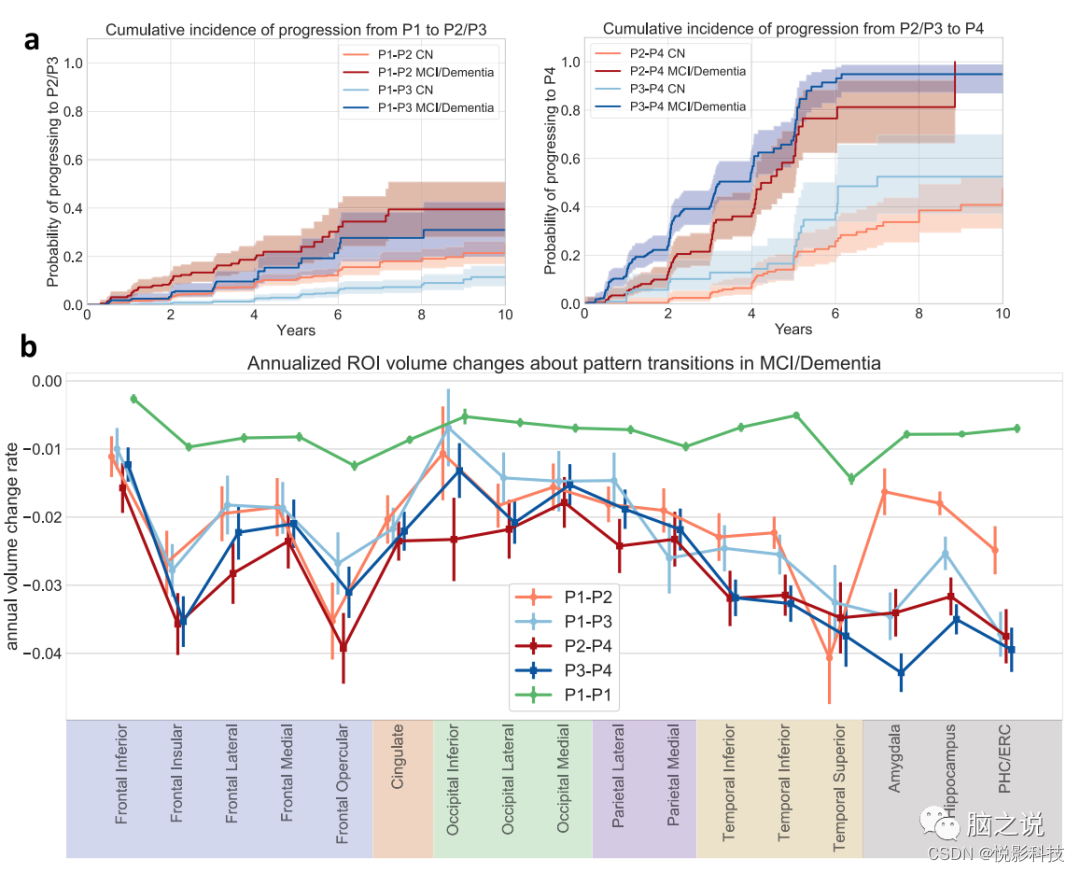
图 4. 纵向模式进展分析。(来自所有2832名ADNI/BLSA参与者的数据)a 模式进展的累积发生率。线条样式表示基线时的诊断。以估计的累积发病曲线为中心显示了95%的置信区间。b沿不同路径选定的转基因区域的年萎缩率。使用模式改变或最后一个随访点(对于稳定的P1参与者(P1-P1))前3年内的数据,并使用随机截距混合效应模型与时间作为固定效应来得出相对于基线体积的年体积变化率。数据表示为时间变量的估计系数±标准误差。
MRI和临床特征
在具有不同主导模式的A+认知障碍参与者中进行MRI和临床特征的统计比较。相对于P2/P3,P4和P1参与者的WML体积分别显着增加和减少(中位
数(第1-3个四分位数)50.6 (39.1-66.1) mm3和34.1 (25.9-49.6) mm3,p < 0.001), 但P2和P3之间没有显着差异(中位数46.3(30.2–59.2)mm3和45.1(33.6–57.2) mm3, p = 0.86)。P3和P4参与者的海马体积相对于总脑容量显着降低(P3和P4的中位数百分比分别为0.54 (0.51-0.56)%和0.52(0.48-0.56)%,而P1的中位数百分比为 0.61(0.58-0.64)%), p < 0.001)。P3参与者的某些特征最高:ApoE ε4等位基因携带率(78%)、tTau(341.1(267.9–446.7))和pTau(34.9(26.1–46.4))水平。具有不同模式的参与者在认知测试分数上存在显着差异,基于A/T状态存在重要差异(图 3c)。无论A/T状态如何,P1参与者在认知领域的表现都要好得多,而P4参与者的表现最差。P2参与者的执行功能表现比P3参与者差,但记忆功能更好。P2 和P3参与者之间的ADNI-EF和ADNI-MEM显着不同(A-T- p = 0.039,A-/T+ p = 0.003,A+/T+ p < 0.001)。对特殊亚组的调查显示了疾病的其他特征。在A+P3参与者中,CN组和受损组的tTau ( p = 0.33) 和pTau ( p = 0.48) 水平相似,表明AD病理变化相当。然而,A+P3 CN参与者的受教育时间明显更长(与A+P3 受损的参与者相比,p = 0.001)、更高的海马体积(p = 0.018)和更少的P3模式概率表达(p = 0.048),这表明更高的认知储备和更少的神经变性可能是保留这个相对较小的群体的认知功能(n = 19)。患有MCI/痴呆的A-T-P1和A+T-P1参与者在认知测试分数或海马体积方面没有显着差异,但A+T+P1参与者确实表现出明显更差的认知表现(图 2c)伴随着海马体更大的萎缩(p = 0.011)和显着降低的P1概率(p < 0.001),表明T的早期不良反应可能与潜在的AD病理学有关,即使没有太多的神经变性存在。
模式类型的纵向进展
图4a中的累积发生率曲线显示,基线时的P1参与者比P3更可能进展到 P2,并且基线时P3的参与者比基线时表达P2的参与者有更高的机会进展到P4。无论基线时的认知诊断如何,这些关系都成立,尽管基线CN的模式进展速度比基线认知障碍患者慢得多。图 4b显示不同进展路径中选定区域的体积变化差异。首先,坚持P1的参与者在所有选定区域的纵向萎缩率要低得多。从P1进展到 P3的参与者表现出更快的内侧颞叶萎缩,而从P1进展到P2的参与者表现出更快的额叶和枕叶萎缩。与P2-P4过渡相关的内侧颞叶萎缩加速。虽然与P4模式一起分类,但在从P2与P3进展的P4参与者之间可以观察到明显的区域萎缩,这让人想起那些早期的模式,并表明P4模式是一种常见的终末期神经变性疾病模式。
预测MRI进展
图5a中的生存曲线说明参与者的基线模式表达与转换为P4的风险相关。基线时的Abeta和pTau状态进一步区分未来转换为P4的风险较高与较低。此外,在基线P1参与者中,他们的基线P2和P3概率预测纵向进展路径和进展速度。使用COX比例风险模型中,作者发现P2的基线概率能够区分从P1到P2的不同事件时间的参与者,并在验证集上实现0.823±0.022的平均一致性指数(C-Index)。使用基线P3概率预测进展到P3的风险的类似分析实现了0.844 ± 0.024 的平均C-Index。因此,P1参与者的基线P2和P3概率可能暗示未来从2年到5 年进展到P2或P3的风险。预测性能在5年风险之后恶化,而且预测沿任一途径进展的最佳阈值随着时间的推移而降低。
临床进展预测(诊断变化)
CN、MCI和痴呆的临床分类提供了有关功能状态的有用信息。图5b中的生存曲线显示,即使在基线时Abeta或pTau状态相似,具有不同模式类型的参与者在临床类别中表现出不同的进展率。MCI到痴呆症进展的差异大于CN到MCI 进展的差异,后者在各组之间发生的频率较低。然而,仅对于基线时P2和P3的参与者,pTau和Abeta状态显着增加了从MCI转变为痴呆症的风险。此外,基线的模式概率具有与SPARE-AD分数相当的预测能力,一种先前验证过的AD 神经变性的预测性生物标志物。两者都显着优于海马体积(在CN-MCI和MCI-痴呆预测中,SPARE-AD vs HV和Pattern vs HV的HV,p < 0.001,图 5c)。此外,与包括APOE基因型、ADAS-cog评分、Abeta和pTau测量在内的其他生物标志物相比,模式概率在预测CN到MCI和MCI到痴呆进展方面表现出相当或显着优越的性能(图 5d,MCI-痴呆预测中的模式与ADAS-Cog比较p = 0.15,模式与所有其他生物标志物之间的比较p < 0.001)。
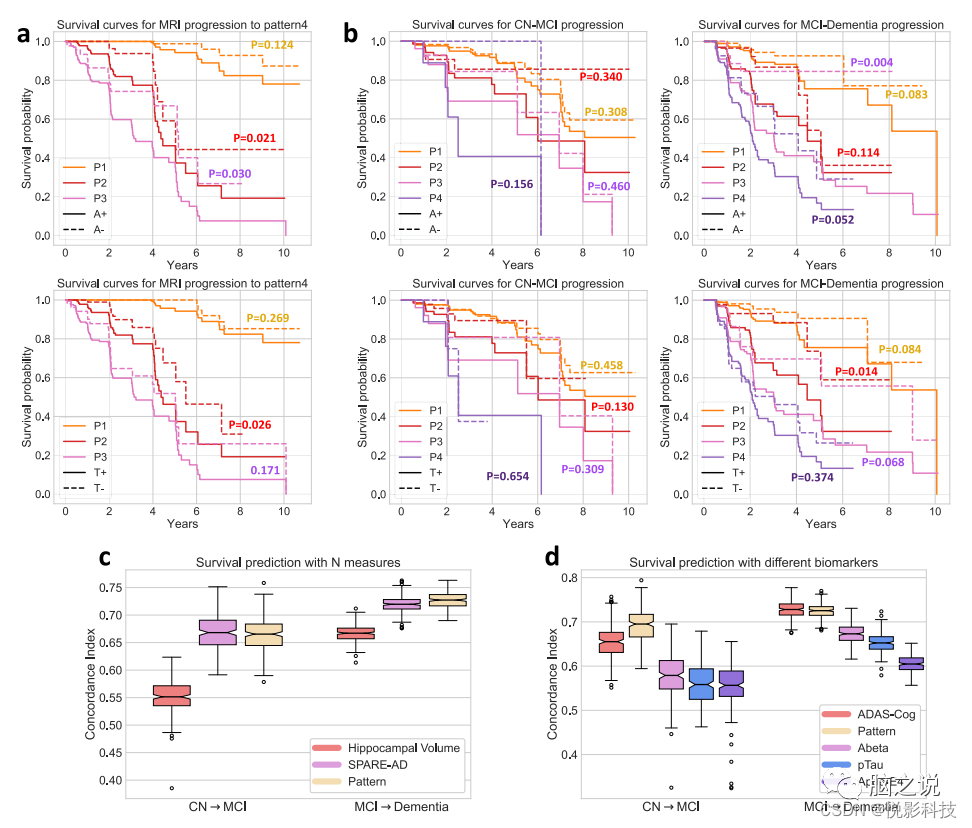
图 5. 模式的预测能力。(来自具有Abeta/pTau测量值的1194名ADNI参与者(a、b、d)和2832名ADNI/BLSA参与者(c)的数据)a神经变性进展至P4的生存曲线;b从CN到 MCI以及从MCI到痴呆的临床诊断进展的生存曲线。对于a和b,生存曲线按初始显性模式和Abeta (A)/pTau (T)状态进行分层;来自对数秩检验的p值表示每个模式内正负Abeta 或pTau状态之间差异的统计显着性;c, d 一致性指数 (C-Index) 的箱须图,用于衡量Cox 比例风险模型在预测临床转换时间(从CN到MCI和MCI到痴呆症)中的性能。不同的生物标志物被用作模型的特征来评估(中心线,中位数;框限,上下四分位数;胡须,1.5×四分位间距;点,异常值)。
临床进展风险综合评分
使用最容易确定的测量值ADAS-Cog作为唯一特征,Cox比例风险模型能够实现平均交叉验证的 C-Index 0.654±0.034预测CN 到MCI 进展和0.728 ± 0.020用于预测MCI到痴呆症的进展。进一步增加源自T1 MRI的模式概率显着提高了两项任务的平均C-Index,分别为0.702 ± 0.042 (p < 0.001)和0.768 ± 0.017 (p < 0.001)。与单独的ADAS-cog相比,单独的模式提供了同等或更好的预测性能。然而,包含来自侵入性脑脊液采样或相对昂贵的PET扫描的Abeta/pTau状态并没有给预测性能带来显着的额外改进(图 6a )。将所有这些生物标志物一起使用,作者可以构建一个综合评分表明临床进展风险,该评分能够预测从MCI 到痴呆的生存时间,在随机拆分的验证集上平均C-Index为0.785 ± 0.016。图 6b显示了一个随机拆分验证集的综合评分分层的生存曲线示例。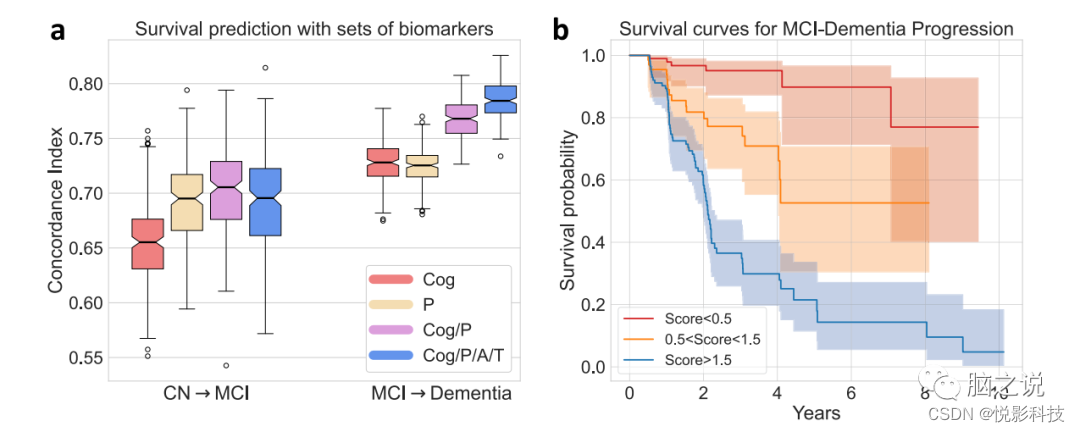
图 6. 使用复合生物标志物预测临床诊断进展。(来自1194名ADNI参与者的Abeta/pTau测量数据)。a生物标志物被连续添加到基于可访问性顺序的特征集中。一致性指数(CI)衡量 Cox比例风险模型在预测临床转换时间(从CN到MCI和MCI到痴呆症)中的性能,不同组的生物标志物被用作模型的特征,以评估其预测能力。(中心线,中位数;框限,上下四分位数;胡须,1.5 倍四分位间距;点,异常值)。b对于一个随机拆分的验证集,按复合评分(A、T、模式、ADAS-Cog以交叉验证方式联合预测结果)分层的生存曲线。以估计的生存曲线为中心显示了95%的置信区间。(A:Abeta;T:pTau,P:Pattern,Cog:ADAS-Cog分数)。
讨论
作者开发了一种深度学习方法,即Smile-GAN,它通过学习生成从认知正常个体的区域体积数据到患者数据的映射,解开病理性神经解剖学异质性并定义神经退行性变的亚型。与无监督方法比较,Smile-GAN的优势在于避免了与疾病无关的混杂变化,从而确定了与病理相关的神经解剖学模式。这源于Smile-GAN 的基本特性,即对从正常解剖结构到病理解剖结构的转换进行聚类,而不是直接对患者数据进行聚类。此外,基于深度学习的Smile-GAN可以轻松处理高维ROI 数据。因此,不需要预处理ROI选择,并且该模型能够完全捕捉所有细分ROI 的变化,并且可以扩展到更小/更多的ROI。此外,与其他半监督方法相比,Smile-GAN没有对数据分布和数据转换线性进行假设,并且在验证实验中发现对轻度、稀疏或重叠的病理模式(此处为神经退行性变)具有鲁棒性。至关重要的是,Smile-GAN给出的模式概率是易于解释的连续生物标志物,反映了各自模式的神经解剖学表达。Smile-GAN的这些优点允许对与病理效应的严重性和异质性相关的模式类型进行多种表征。
Smile-GAN本质上不模拟疾病阶段,也不假设模式之间的任何进展途径。可以在对每个已识别模式的表达程度或它们的线性或非线性组合进行二级分析时推断分期。在ADNI/BLSA中,大多数人同时表达多种模式,这反映在各种模式概率的大小上,这一观察类似于Zhang等人的发现。此功能允许调查基于模式的阶段和感兴趣的临床结果之间的复杂和非线性关系,这些关系可能因结果而异(例如,从各种认知或临床测量到为临床试验招募提供信息的分期估计)。
将Smile-GAN应用于来自富含AD病理学的样本的MRI数据,确定了在整个AD谱系参与者中表达的4种区域性脑萎缩模式,这些模式在验证实验(包括排列测试)中具有高度可重复性。这些模式的范围从轻度到晚期萎缩,并定义了两种进展途径。一种通路,这里称为P1-3-4通路,显示了AD典型的内侧颞叶早期萎缩。第二个通路,P1-2-4,显示早期弥漫性轻度皮质萎缩,MTL保留,这是 AD的不太典型的模式。两种途径的末期都是晚期萎缩模式P4。这种四模式系统与其他基于神经影像的聚类研究有相似之处,包括时间和皮质主要模式的识别。Smile-GAN模式暂时对应于AD的病理识别亚型:边缘为主、匹配P3、海马保留、匹配P2和典型AD(混合P2-P3)。还有另一种可能的皮层下萎缩亚型,之前由其他几种基于MRI的无监督聚类算法使用ADNI 数据确定,但Smile-GAN 没有明确识别。Smile-GAN没有识别出皮层下模式有几个原因。首先,正如 Zhang 等人所观察到的那样,皮层下模式可以与基于协调和聚类方法和特定训练样本的时间模式合并。其次,归因于皮层下萎缩的部分变异性可能与疾病无关,导致信号不足以单独聚集为不同的模式,这种可能性可能由缺乏该亚型的病理学证据和皮层下ROI萎缩稀少支持在患者组中。
四种Smile-GAN模式具有临床意义。模式成员与认知测试表现的差异相关,P2表现出相对较多的执行功能障碍,P3表现出更大的记忆障碍,而P4表现出跨领域的最差表现。这些模式也对进展的速度和方向有影响,早期模式特征预测未来的神经变性模式,模式特征预测从CN到MCI和MCI到痴呆的临床进展。至关重要的是,模式表达是临床进展的最重要预测因子,显示出与其他N测量和生物标志物相当或更强的预测能力(图 5)。协同作用下,模式表达、A、T 和 ADAS-Cog在个体基础上提供了出色的交叉验证临床进展预测(图 6b),强调了这种组合预测指数对患者管理、临床试验招募和用于评估治疗反应。
虽然模式在区域特异性和严重性方面相对不同,但潜在的病理生理学更为复杂。P1表明不存在明显的神经变性。然而,相当数量的P1模式的参与者(N = 306)仍有客观的认知障碍,并伴有MCI,甚至有少数痴呆病例,甚至有一些痴呆病例,无论有无淀粉样蛋白和tau沉积的证据。这些参与者可能减少了认知储备和/或对MCI/痴呆的非神经退行性贡献。P2组表现出轻度弥漫性萎缩,可能包含多种轻度或早期病理,包括AD的海马保留/皮质表现和其他早期神经退行性过程或与慢性全身性疾病相关的萎缩,部分由淀粉样蛋白阴性 P2 参与者证明. 然而,P2并没有过多地富含血管疾病,这是原发性神经变性疾病的常见合并症,至少根据P2-P3-P4相对相似的WML体积测量。无论病因如何,虽然P3主要是丰富的ADNI样本中的早期典型AD,但这也可能包括其他病理,例如边缘为主的年龄相关TDP-43脑病 (LATE)。P4似乎是晚期或“末期”神经退行性变模式的复合体。虽然完全是晚期AD的典型特征,但在没有淀粉样蛋白或 tau沉积的认知能力下降的参与者中也可以看到这种模式,这表明在多种疾病中广泛存在脑萎缩的晚期相似性。
随着AT(N)框架的使用越来越多,这些模式提供了一种将神经变性量化为几个信息类别而不是二元测量的方法。将神经变性分类为无(P1)、早期皮质 (P2)、时间主导(P3)或高级(P4)提供了重要的表型信息,同时保留了二元AT(N)框架的大部分简单性。与A/T状态一起,模式表达的动态变化显示了疾病的严重程度,并确定了具有不同平衡的AD和非AD病理学的合理的、不同规模的亚组(图 7)。这些组可用于丰富临床试验的典型AD病理学,减少确定某些生物标志物的需要,并确定有趣的亚组进行重点评估,例如恢复力的遗传因素。例如,要招募具有早期典型AD神经退行性变的组,可以首先选择MRI上具有P3模式的那些(本研究样本中为25.3% A+T+的组)并仅在该组中确定A/T生物标志物。
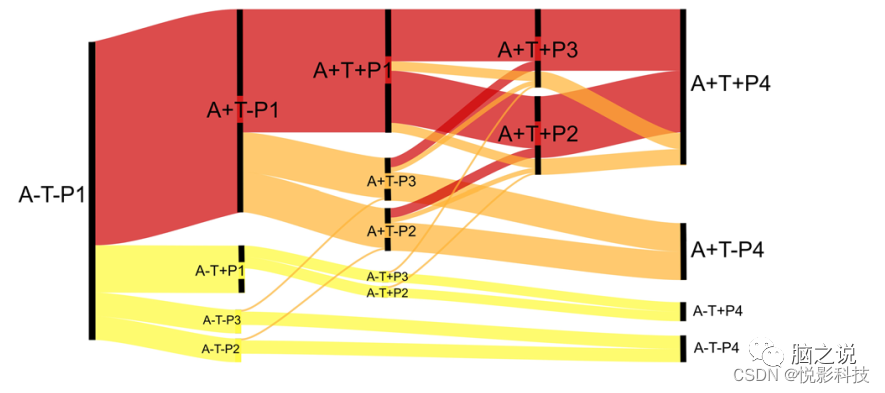
图 7. 模式路径对ATN框架的影响的假设流程图。级联的生物标志物可以遵循典型的AD途径,该途径在ADNI样本中最具代表性(红色)。模式与淀粉样蛋白/tau状态的关系确定了另一个存在AD病理学和显着甚至显性共病理学(橙色)的大组以及疑似非AD病理学的组(黄色)。这些途径还表明,某些典型的AD神经退行性表型在某些情况下可能是由共病理学驱动的。例如,A+T+节点是AD的典型节点;然而,有几种潜在的路径(橙色),其中共病理学可能是神经退行性模式的主要原因。路径厚度估计通过ADNI中的节点的近似通量。该模型基于A/T/P类别中的横截面数据分布以及事件以特定顺序发生的假设(A-→A+;T-→T+;P1→P2→P4和P1→P3→P4)。
Smile-GAN模式方法有几个优点。它捕获了数量很少的生物学相关区域萎缩模式,在不需要显着复杂性的情况下提供有关神经退行性变的有意义的顶级细节,同时保持定量模式概率信息。Smile-GAN方法是一种数据驱动的方法,可以应用于从神经影像以外的数据中提取的特征,可能能够根据从正常组到患者组的任何选定的疾病相关特征变化有效地对患者进行聚类。因此,它可以推广到任何在成像或其他生物医学数据中具有可重复变化模式的疾病和障碍,包括但不限于其他神经变性疾病和神经精神疾病。虽然训练每个模型的时间和计算要求适中,主要取决于特征的数量,但训练过程只执行一次,随后使用现有模型计算单个模式分数的速度很快。该方法和作者的实施存在局限性。首先,对照组的选择对结果模式产生关键影响,因为根据设计,对照组中常见的任何变化都不会被明显隔离。例如,这可能是血管疾病分布在多种模式中的原因。类似地,模型可能无法清楚地学习稀有和/或细微的萎缩模式,如模拟实验所示。更大和更多样化的训练数据可能允许在AD连续体中识别更多模式类型。将参与者分配到组的阈值可能会受益于针对特定假设量身定制的优化。本研究中四模式模型的性能是使用ADNI 和BLSA研究的数据得出和评估的,这两个研究分别具有高和低的AD患病率,以及相对较低的非AD神经退行性变患病率。尚未评估将该模型直接应用于患有混合性神经变性疾病的记忆中心人群。最后,Smile-GAN 模型目前仅应用于源自MRI图像的ROI体积数据,因此可能无法捕获不符合解剖ROI的更细微的模式。Smile-GAN模型架构可灵活用于较小的ROI分区或基于体素的分析以及非结构MRI和非成像数据。将当前框架扩展到此类其他类型的数据是未来发展的方向。
使用具有广义对抗网络的半监督聚类识别的模式提供了有关整个AD谱中神经退行性变的严重性和分布的有用信息。基线模式可预测未来的神经变性模式以及临床进展为 MCI 和痴呆。这些模式可以增强对认知能力下降的参与者和患者的研究和临床评估,并有助于对脑部疾病和障碍进行维度表征。
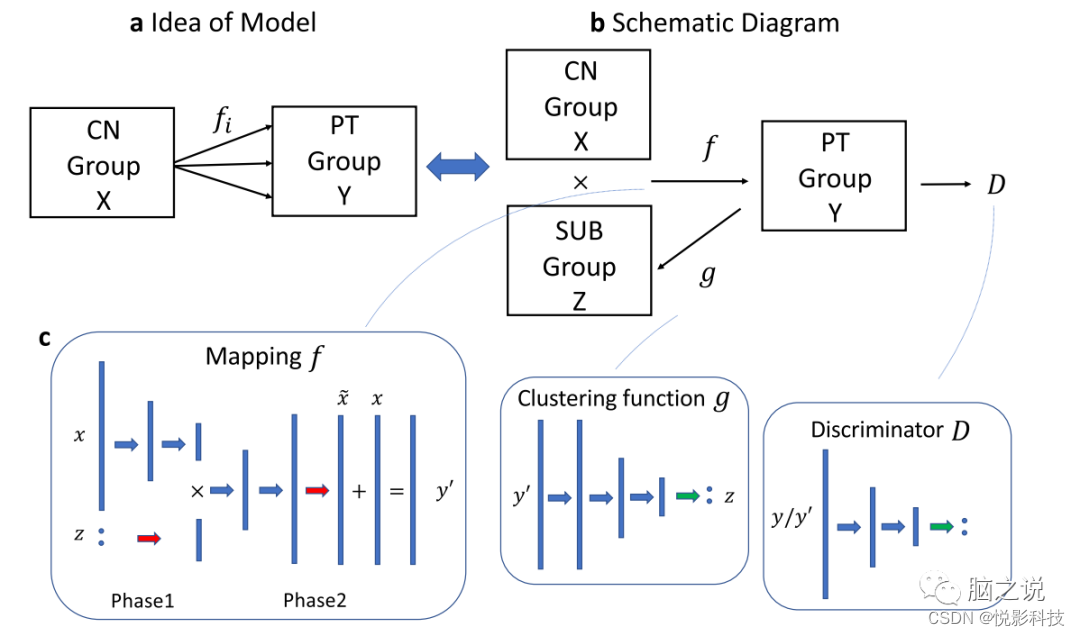
图 8. 示意图和网络架构。a Smile-GAN的总体思路。该模型的目的是学习从CN组到PT组的几个映射 bSmile-GAN的示意图。该模型的想法是通过学习一个从两组X×Z联合到Y的映射来实现的,同时学习另一个函数g:Y→Z。c 三个函数的网络结构:蓝色箭头代表一个线性变换后有一个漏整流线性单元函数,绿色箭头代表一个线性变换后有一个softmax函数,红色箭头代表只有一个线性变换。
方法
Smile-GAN 模型
Smile-GAN是一种生成对抗网络(GAN)架构,用于根据多变量差异(在本案例中为来自MRI的区域体积)将一组(患者)聚类到参考组(健康对照)。Smile-GAN的一般结构如图8所示。综上所述,该模型的主要概念是学习从CN组X(或者称为域X:CN数据集)到患者(PT)组Y(或者称为域Y:PT数据集)的一对多映射。这个想法相当于学习了一个映射函数f:X×Z→Y,它从真实的CN数据和采样的子类型变量z中生成合成的PT数据y′=f(x,z),同时强制PT数据和合成的PT数据之间不可区分。简单地说,给定一个相同的亚型变量z的值,映射f(⋅)产生的图像数据与类似亚型混合的病人的数据相匹配。这里,Z={α:∑Mi=1αi=1},被称为亚型(SUB)组(也称为SUB域),是一类具有M维的向量(在作者的实验中发现M=4是最佳的)。作者将上述变量的分布分别表示为x∼pCN、y∼pPT、y′∼pf、z∼pSub。变量z独立于x,从Z组的一个子类中取值,可以编码为一个单枪匹马的向量,其值1以相等的概率放在任何位置(即1/M)。除了映射函数外,还引入了一个对抗性鉴别器D,以区分真实的PT数据y和合成的PT数据y′,从而确保映射f产生的图像数据与真实的病人数据无法区分。
一些函数有可能实现分布上的相等,这就很难保证模型学到的映射与基本的病理进展密切相关。此外,在训练过程中,神经网络支持的映射函数往往会忽略子变量z的存在。因此,假设真实的PT变量y=h(x,z)有一个真正的底层函数,Smile-GAN旨在通过三种类型的正则化来约束函数类别,从而提高映射函数f近似于真正的底层函数h:(1)作者鼓励稀疏转换,(2)强制执行函数的Lipschitz连续性,(3)向模型结构引入另一个函数g:Y→Z。后者是算法对数据进行聚类的关键部分,因为它要求映射函数在Y组中识别足够不同的成像模式,这将使反映射g(⋅)能够估计PT组中正确的子类型。
Smile-GAN的目标是对抗性损失和正则化条款的结合。首先,对抗性损失旨在将合成的PT数据的分布pf与真实的PT数据的分布pPT相匹配。第二,正则化条款包括变化损失和聚类损失,两者都用于限制学习f的函数空间。
研究和参与者
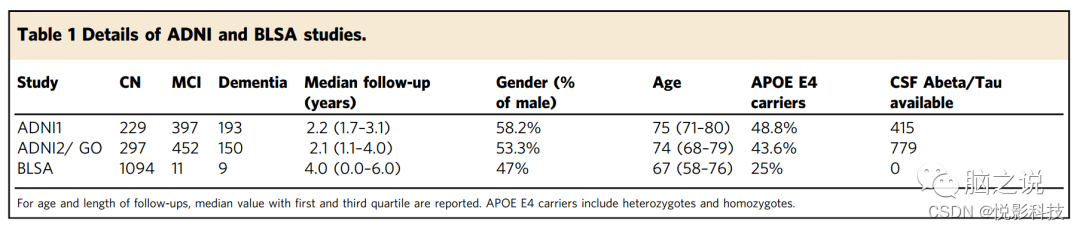
阿尔茨海默病神经影像学倡议 (ADNI, http://www.adni-info.org/ ) 研究是一项公私合作的纵向队列研究,通过4个阶段(ADNI1 , ADNIGO, ADNI2)招募了认知正常、MCI和AD参与者。ADNI已获得纵向MRI、脑脊液(CSF)生物标志物和认知测试。自1993年以来,巴尔的摩老龄化纵向研究(神经影像学子研究)一直在跟踪那些在影像学和认知检查中登记时认知正常的参与者。总共包括 1718名ADNI参与者(819名ADNI1和899名ADNIGO/ADNI2)和1114名 BLSA参与者被纳入该研究中。详细的入选标准信息可参见Peterson等的ADNI和Resnick等的BLSA。两项研究的详细信息,包括基线时归类为CN/MCI/痴呆的人数、具有CSF Abeta/Tau生物标志物的参与者人数、随访时间、年龄、性别、APOE基因型,均包含在表1中。参与者对ADNI和BLSA研究提供了书面知情同意书。这项研究的方案得到了宾夕法尼亚大学机构审查委员会的批准。
表1. ADNI和BLSA研究的详情
MRI数据采集和处理
从上面介绍的ADNI和BLSA研究中获得了1.5T和3T MRI数据。应用全自动流水线处理T1结构MRI。每个参与者的T1加权扫描首先针对强度不均匀性进行校正。应用多图集颅骨剥离算法去除颅外材料。对于ADNI研究,使用多图集标签融合方法在灰质(GM,119个ROI)、白质(WM,20个ROI)和心室(6个ROI)中识别出145个解剖感兴趣区域(ROI)。对于BLSA 研究,该方法与统一的特定采集图集相结合导出相同的145个ROI。对145个ROI的区域体积应用阶段级横截面协调以消除场地影响。对于疾病模式的可视化,组织密度图,称为RAVENS(在标准化空间中检查的体积的区域分析)计算如下。单个图像首先注册到单个受试者的大脑模板并分割成GM和WM组织。RAVENS地图对每种组织类型进行本地和单独编码,在配准期间观察到的体积变化。
数据分离和准备
预处理后,选择来自ADNI2/GO参与者的297名CN和602名认知障碍参与者的基线ROI数据作为模型训练和验证的发现集。来自ADNI和BLSA的所有参与者的后续访问的纵向ROI数据用于进一步的临床分析,包括那些基线数据被用于模型训练的参与者和那些完全独立于发现集的人。对于需要测量CSF Abeta/pTau的分析,仅包括具有这两种生物标志物的ADNI参与者。否则,将所有来自ADNI和BLSA研究的参与者纳入分析。
在用作Smile-GAN模型的特征之前,对ROI体积进行了残差和方差归一化。为了纠正年龄和性别影响,同时保持与疾病相关的神经解剖学变异,作者使用线性回归模型估计了297名CN参与者的ROI特定的年龄和性别关联。然后,所有横截面和纵向数据都通过年龄和性别效应进行残差。然后,针对发现集中的 297名CN参与者进一步标准化所有ROI体积,以确保每个ROI的CN参与者的平均值为1和标准差为0.1。
认知、临床、脑脊液生物标志物和遗传数据
作者使用了额外的临床、生物流体和遗传变量,包括由ADNI提供的脑脊液淀粉样蛋白和tau生物标志物、APOE基因型和认知测试分数。这些措施是从 LONI网站下载的。来自ADNI的1194名参与者在基线时对β-淀粉样蛋白、总 tau和磷酸化tau进行了CSF测量,其中包括383名CN、578名MCI和233名痴呆症;BLSA参与者没有A/T生物标志物,因此被排除在基于这些措施的分析之外。Hansson等人描述了CSF量化的详细方法。基于ß-淀粉样蛋白测量的淀粉样蛋白状态和基于磷酸化tau测量的tau状态的截止值先前已定义并用于将参与者分类为脑淀粉样蛋白和tau沉积的阳性或阴性。Tau测量值也表示为连续变量。多个领域的综合认知评分先前已在ADNI队列中得到验证。记忆复合(ADNI-MEM)模型基于Rey听觉语言学习测试、阿尔茨海默病评估量表-认知分量表 (ADAS-Cog)和简易精神状态检查(MMSE)中的组件。执行功能复合(ADNI-EF) 模型基于动植物类别流畅性、追踪A和B、数字向后跨度、修订后的韦氏成人智力量表中的数字符号替换以及圆圈、符号、数字、手和时钟绘图任务中的时间项。语言复合(ADNI-LAN)模型使用动物和植物类别流利度、波士顿命名总数、MMSE 语言元素、ADAS-Cog的以下命令/对象命名/概念实践和蒙特利尔认知评估 (MoCA)语言元素,包括字母流利度、命名和重复任务)。
白质病变(WML)体积是从ADNI和BLSA计算的,使用不均匀性校正和共同配准的FLAIR和T1加权图像以及基于U-Net架构的基于深度学习的分割方法,卷积网络中的层被Inception ResNet架构取代。该模型是使用单独的训练集进行训练的,该训练集具有人工验证的WML分割。WML体积首先是立方根的。然后对它们应用相级横截面协调以减少站点效应。
模式成员资格和概率分配
Smile-GAN模型为每个参与者分配M个概率值,每个概率对应一种模式类型,并且M个概率之和为1。基于M个概率值,作者可以进一步将每个参与者分配给主要模式类型,由下式确定最大概率。基于聚类再现性或稳定性,在交叉验证(CV)过程中选择最佳M。具体来说,对M=3到5的重复保持CV进行了10次折叠。对于每一倍,作者随机遗漏了20%的发现集以增加可变性。值得注意的是,M = 2通常将数据分层为轻度和重度萎缩模式,这在临床上并不有趣。作者使用调整兰德指数(ARI)来量化10个折叠/模型的聚类稳定性。ARI是随机索引的机会版本的校正,对于两个随机分区等于0,因此,它被认为是测量聚类结果重叠的好选择。在M = 4时达到最高的平均成对ARI,0.48 ± 0.08,对于M = 3,ARI = 0.30 ± 0.12,对于M = 5,ARI = 0.33 ± 0.07。置换实验证明了Smile- M = 3-5的GAN模式,由ARI 测量。总之,这些数据表明M = 4产生最佳聚类数。
在M = 4的情况下,作者使用发现集中的所有可用数据重新运行Smile-GAN 30次,训练后的模型将用于外部验证和分析。为了在30个实验中找到集群分配之间的最佳对应关系,作者计算了每个结果模型的平均成对ARI值。选择具有最高ARI的模型作为模板,并对所有其他模型学习的模式类型进行重新排序,使其聚类结果与模板的聚类结果达到最高的重叠。重新排序后,将所有30个模型中每个模式的平均概率作为每个参与者对应模式的概率。
统计分析
为了可视化四种模式的大脑特征,作者利用发现集中MCI/痴呆症参与者的所有横截面数据,并使用体素组织密度通过AFNI 3dttest进行体素组比较(即CN 与每种模式)。为了访问模式分配的纵向进展轨迹,作者将概率大于0.5的参与者对四种模式中的每一种进行分组。然后,作者通过计算在给定时间间隔(即X 年-X+1年)内具有可用数据的组内所有人的P1-P4的模式概率,比较了四组中每一组的模式概率如何随时间变化。那些在选定的时间间隔内有一个以上的数据点的人只通过所有这些访问的平均概率贡献一次。人口统计学变量、APOE基因型、CSF生物标志物水平、认知测试分数、WML体积和模式概率在模式类型之间和模式类型内进行了比较。只有来自ADNI研究的参与者,其Abeta/pTau状态在基线时可用,才被纳入比较。对于分类变量,卡方检验用于识别亚组之间的差异。对于其他定量变量,进行单向方差分析以进行组比较。通过在线python包、statsmodels 0.8.0、SciPy 1.6.3、NumPy 1.16.6和pandas 0.21.0进行统计分析。
为了评估从P1转换为P2或P3的风险,作者进行了时间-事件生存分析以评估风险模式转换。特别是,作者将P2和P3视为竞争事件,并使用Aalen-Johansen 估计器生成P1到P2/P3进展的累积发生率曲线。对于与模式进展和诊断转换相对应的所有其他累积发病曲线和生存曲线,作者应用了非参数Kaplan-Meier估计量,并使用对数秩检验来比较组间生存分布的差异。
对于所有生存分析,参与者在基线时被分配到一种模式或仅在相应模式概率大于0.5时标记为进展到一种模式。一些参与者在基线时以任何模式都没有达到这个阈值,因此被丢弃以避免分析中的噪音。所有生存分析均通过在线python lifelines 0.25.7进行。
评估模式的预测能力
作者进一步进行了分析,以评估基线模式概率在预测未来模式变化中的预测能力。此外,作者将它们与其他神经变性测量(N测量)和临床生物标志物进行了比较,以预测诊断转换。
对于模式进展预测,作者选择了所有940名进行纵向随访且基线P1 > 0.7的参与者,以避免琐碎的预测任务。首先,使用以基线P2或P3概率为唯一特征的 Cox比例风险模型分别预测从P1到P2或P1到P3的生存曲线。作者运行了两次交叉验证100次,并得出了验证集的一致性指数。其次,为了预测P1参与者在特定时间点X的模式进展风险和进展路径,作者直接使用基线的P2概率和 P3概率作为风险指标,而无需进一步拟合任何其他模型。对于从2年到8年的每次X,作者生成了一个二元指标,0表示直到T才进展到P2,1表示在T之前已经进展到P2,并直接使用基线P2概率来区分这两组。对P3也进行了完全相同的过程。计算了不同时间X的P2和P3的接收者操作员特征曲线下面积(AUC) 值。报告了两种不同进阶途径的最佳鉴别阈值,其中真阳性率(TP)加上假阳性率(FP) = 1。
为了预测临床诊断变化,作者选择了1178名CN参与者和921名在基线时被归类为MCI的参与者进行了纵向随访。首先,为了将Patterns与其他N测量值进行比较,作者再次利用具有不同N测量值的Cox比例风险模型作为预测CN-MCI 和MCI-痴呆生存曲线的特征。两次交叉验证运行100次以得出验证集的一致性指数。然后,为了比较Pattern、Abeta、pTau、APOE 基因型和ADAS-Cog 的预后能力,作者将样本减少到拥有这些生物标志物的380名CN和568名MCI 参与者。每个生物标志物都被独立用作训练模型的唯一特征。
生物标志物选择和综合评分构建
最后,作者评估了上述不同生物标志物组合的预测能力。按照可访问性的顺序,ADAS-Cog、源自T1 MRI的模式概率、源自PET扫描的Abeta/pTau依次添加到用于训练Cox比例风险模型的特征集中,并实施了与本文介绍的相同的实验程序上一节。可以使用上面介绍的所有生物标志物得出指示临床进展风险的综合评分。使用基线的Pattern概率/Abeta/pTau/ADAS 分数作为特征,将经过训练的Cox比例风险模型应用于验证集,以得出部分风险作为综合分数。
参考文献:A deep learning framework identifies dimensional
representations of Alzheimer’s Disease from brain structure
版权归原作者 悦影科技 所有, 如有侵权,请联系我们删除。