
背景
深度学习在计算机视觉和自然语言处理(NLP)等领域的“有效性”依赖于深度神经网络利用不断增长的计算量、数据和模型容量的能力。大多数SOTA的NLP、CV大模型都是基于一个小集合内的大型预训练模型改编而成的,通过自监督预训练可以很成功地从大型数据集中合成信息,执行各种下游任务时几乎没有甚至根本没有微调。因此,大规模的模型和数据集扩展可能将是深度学习在科学领域取得巨大成功的先决条件。
AlphaFold、Open Catalyst Project和ChemBERTa等最近的工作表明,更大的数据集和模型、预训练和自监督学习,这些计算机视觉和NLP中的关键要素,都为深度学习在化学领域的应用解锁了新的可能性。
然而,与计算机视觉和NLP不同的是,规模化化学深度学习网络的途径和收益尚不明确:一方面,化学深度学习可以结合领域先验知识,这可能可以缓解紧迫的资源需求;另一方面,由于化学空间和分子机器学习任务的异质性和复杂性,想要训练出在各种下游任务上均表现良好的、通用且鲁棒的模型会是一个不小的挑战。然而,化学空间的巨大性和下游任务的异质性使得化学领域的大模型非常适合无标签的多模态数据集。与此同时,最近的研究发现,neural scaling law在多个数量级的模型大小、数据集大小以及计算量上都改善了模型性能的表征。但这些实验都需要大量的计算资源,并且依赖于特定领域的模型训练程序,这些程序又并不适用于传统深度学习应用领域之外的领域。并且,大模型的开发部署成本高昂,neural scaling行为规律的研究也依赖于昂贵的超参数优化及实验。
借鉴CV和NLP领域利用加速神经架构搜索和超参数传递的技术,如TSE、μTransfer,可以加速深度学习大模型开发的思路,MIT的学者Nathan C. Frey及其团队提出,要研究化学深度学习模型的在不同规模下的能力,需要找到可行并且规范的方法来加速超参数转移和表征神经尺度。
他们开发了深度学习化学大模型的scaling策略,通过在多个数量级上改变模型和数据集大小来研究大型化学模型中的scaling行为。介绍了具有超过10亿个参数的chemGPT模型,在多达1000万个数据点的数据集上进行预训练,并研究了用于生成式化学建模的大语言模型LLM和用于学习原子间势的图神经网络。此外,该研究团队还探索了物理先验和尺度之间的相互作用,并发现了化学中语言模型的经验scaling关系,最终得到,在实验范围内的最大数据集上,scaling指数β为0.17,对于等变图神经网络原子间势,scaling指数β为0.26。
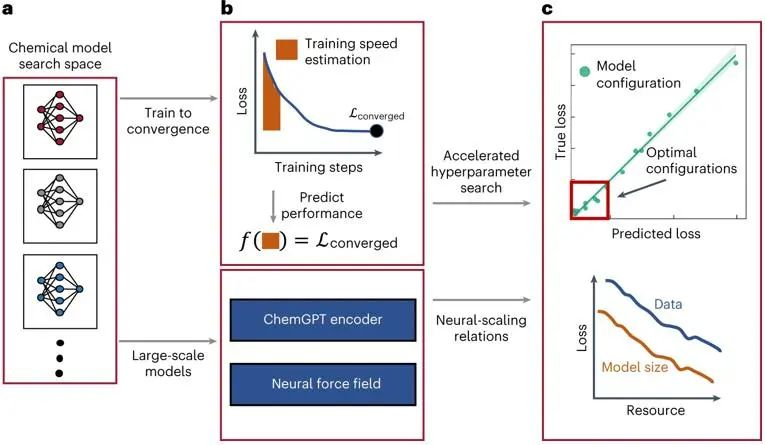
图1. 深度学习化学模型的scaling关系探究
1、****方法
对于PEC介质,表面等效电流满足电场积分方程(EFIE)和磁场积分方程(MFIE):
1.1 Neural scaling法则
对具有足够模型参数和/或数据收敛的LLM和CV模型而言,其性能随模型参数量、数据集大小及计算量的经验scaling关系可以由式1给出。
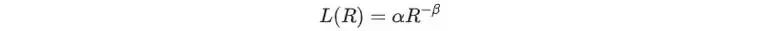
式1
其中α为scaling系数,R指资源信息量,包括模型参数量,数据集大小以及计算量,β为scaling指数,表征此幂律的斜率,指示模型相对R的scaling效率。对于固定的数据预算,缩放指数量化了由于模型大小增加而带来的损失改善。β值越大,对应的斜率越陡,随着数据/模型大小的增加,性能越好。需要注意的是,此经验公式不适用于分辨率受限的情况,即数据集足够大但数据集不够大,反之亦然。从神经尺度关系中识别这些分辨率受限的区域,可以让我们大致理解模型损失改善是受限于数据可用性还是受限于模型容量。
1.2 化学大语言模型
化学图谱天然地可以用字符串简单表示,因此时序模型是处理化学数据的天然选择。观察到基于transformer的模型预训练损失可以通过数据集或者模型体量的增加而得到明显的改善,该团队为化学设计了一个名为ChemGPT的生成式大语言模型,以研究数据集和模型大小对预训练损失的影响。ChemGPT是一个基于GPT-Neo的GPT3风格的模型,带有用于描述分子的自引用嵌入字符串SELFIES的tokenizer。对于化学语言建模,一组分子(x1、x2、...、xn)被表示为每个分子的符号序列(s1、s2、...、sn),给定一个序列,p(x)的概率可以因式分解为各个分子的条件概率的乘积。ChemGPT使用具有自注意力机制的transformer架构来计算条件概率,估计p(x)并从中采样以生成新的分子。该团队的ChemGPT具有多达10亿非嵌入参数,在包含了多达1000万个分子的PubChem数据库上进行预训练,相比于传统的生成式化学模型,其规模大大提升了。
1.3 GNN力场
对于大多数化学领域的下游任务,分子构型和三维结构信息是必要的。该工作使用GNN接入分子中原子的坐标信息,并预测给定分子形貌的能量,微分获得能量守恒的原子力场。
1.4 Training Performance Estimation(TPE)
由于模型超参数(包括学习率和batch大小)对于实现最优损失至关重要,但在不同领域和模型/数据集大小之间不可转移,因此我们需要有效的策略来在深度化学模型中实现可扩展的超参数优化(HPO)。为了在计算资源限制下实现深度化学模型的高效scaling,我们引入了TPE,这是TSE的一个延伸,它降低了HPO的计算成本,发现哪些超参数在新领域应用中最重要,以及要研究哪些超参数,通过在训练过程中自动提前停止来加速HPO。用来加速化学语言模型和GNN原子间势的模型选择。
2、****结果
物理信息监督残差学习被提出作为电磁建模的通用深度学习框架 [2]。它应用深度神经网络(DNNs)来通过残差学习矩阵方程中未知量的更新函数,用于迭代地修改未知量,直到收敛。在残差学习的框架下,第k步迭代的更新方程如下:
2.1 加速超参数优化
(1)TPE加速ChemGPT的超参数优化
图2展示了在MOSES数据集的200万个分子上训练的ChemGPT模型的训练性能估计(TPE)结果。MOSES比PubChem更小,并且是训练化学生成模型的代表数据集。这里使用MOSES来演示如何使用TPE快速发现化学LLM(如ChemGPT)的最佳设置。图2中用20%的数据验证TPE的可用性,显示了50个epoch后的真实损失与仅10个epoch后使用TPE的预测损失,线性回归的R^2=0.98。此过程在新的数据集上比较容易被重现,并且可以加速HPO。
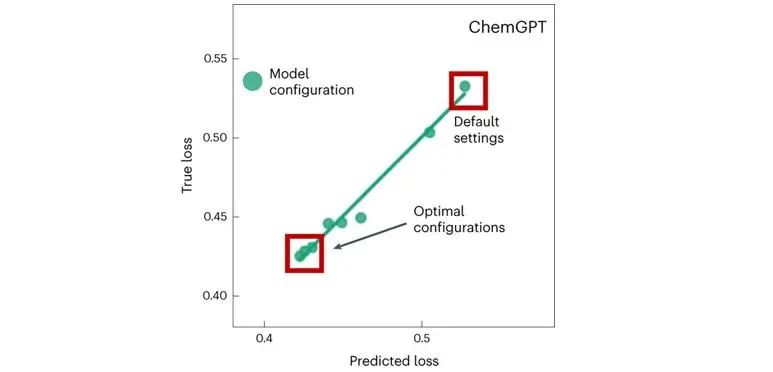
图2. 使用TPE在训练早期识别最佳模型,并停止非最优模型的训练,以节省80%+的总计算消耗
(2)TPE加速GNN超参数优化
TPE对于GNN的表现同样出色。重复上述过程,使用总训练预算的20%,改变学习率和批量大小,用于SchNet、PaiNN和SpookyNet,在数据集MD17上训练。SchNet和PaiNN的TPE实现了出色的预测能力(图3)。发现TPE的效果与使用全部训练预算的模型损失方差息息相关,这也表明了合适的HPO的重要性。
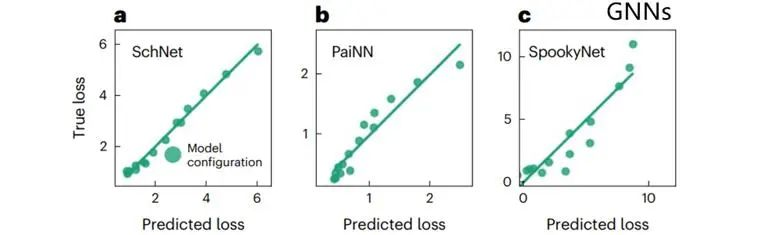
图3. TPE对于GNN的表现同样出色
2. Neural Scaling公式的拟合
在neural scaling的介绍中提到的“分辨率受限”的情况,即模型和数据集规模不适配。从scaling关系中识别这些分辨率受限的区域,可以让我们大致理解模型损失改善是受限于数据可用性还是受限于模型容量。对于固定的数据预算,scaling指数量化了由于模型大小增加而带来的损失改善。根据数据集的大小,不同范围的模型大小可以看到类似幂律的缩放行为。Scaling公式拟合在图像上可表示为损失与模型大小在对数-对数图上的近似直线拟合。指数β值越大,对应的斜率越陡,随着数据/模型大小的增加,性能越好。图4给出了给定数据集大小,损失随着模型参数量的变化,且通过不同数据集大小,β值的大小演示了数据集大小对模型性能的影响。而拟合直线的断裂也标示着分辨率受限区域的存在。
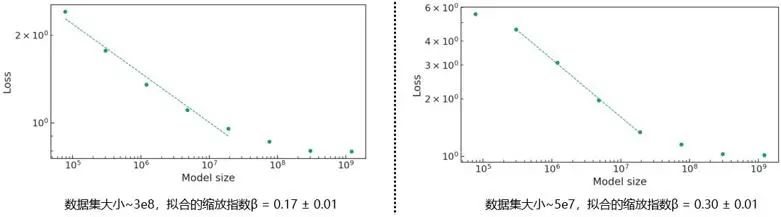
图4. 拟合ChemGPT中的neural scaling公式
在scaling法则成立范围内,模型性能与数据集大小,模型大小及容量是具有单调性的(图5左)。表明在一定量级内,想要提升模型性能,可以通过简单的增加数据集或者扩大模型得到提升。与此同时,对于GNNs/NFFs,低容量模型的收益随着数据集大小的增加而递减,而高容量模型则随着数据集大小的增加而快速改善(图5右)。因此,扩展模型和数据集大小的好处应该与增加的计算成本进行平衡,以找到计算和数据效率最高的改进机会。
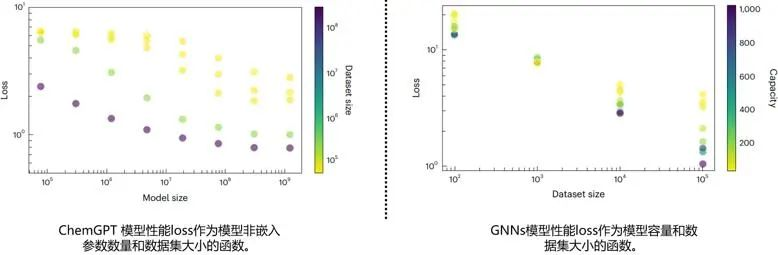
图5. 化学GPT模型性能的神经缩放(验证损失)是模型(非嵌入参数的数量)和数据集(令牌的数量)大小的函数
3、****感想
该研究工作的核心贡献是发现了用于缩放化学领域大语言模型和GNN原子间势的策略(neural scaling law),量化了模型损失如何在多个数量级上取决于模型规模和数据集大小。并且发现对于化学领域大语言模型和NFF,在模型大小、数据集大小或计算方面都没有饱和模型损失。最后,基于物理的先验对scaling行为的影响提供了丰富的描述,说明了将物理、已知的经验关系和其他形式的知识纳入机器学习框架是如何影响学习质量和效率的。
研究结果为科学深度学习中的尺度研究提供了动力和实践指导,并为大规模和物理深度学习的交叉点提供了许多富有成效的新研究方向。这些结果可以优化分配计算和数据预算,以实现最大效率的模型损失改进,并使可扩展的科学深度学习更适合更广泛的研究领域。
参考文献
[1]Frey, N.C., Soklaski, R., Axelrod, S. et al. Neural scaling of deep chemical models. Nat Mach Intell 5, 1297–1305 (2023). https://doi.org/10.1038/s42256-023-00740-3
[2]https://github.com/ncfrey/litmatter.git
[3]https://github.com/learningmatter-mit/NeuralForceField.git
[4]https://github.com/datamol-io/molfeat.git
[5]https://github.com/coleygroup/rogi-xd.git
往期回顾
MindSpore AI科学计算系列 | GNN-MoM基于昇思MindSpore Elec的图残差学习电磁求解器
MindSpore AI科学计算系列 | VAE基于MindSpore Elec的特征域MT反演,提升大地电磁反演的精度和分辨率
MindSpore AI科学计算系列 | Allegro提升昇思分子势能预测准确性,将上线MindSpore Chemistry
MindSpore AI科学计算系列 | 周期性图Transformer提升MindSpore模型对晶体性质的预测
版权归原作者 昇思MindSpore 所有, 如有侵权,请联系我们删除。