
Deep learning methods for molecular representation and property prediction
总结
随着人工智能方法的进步,计算机辅助药物设计(CADD)近年来发展迅速。有效的分子表示和准确的性质预测是CADD工作流程中的关键任务。在这篇综述中,作者总结了当前深度学习方法在分子表示和性质预测方面的应用。作者根据分子数据的格式(1D、2D和3D)对深度学习方法进行了分类。此外,文中还讨论了一些常见的深度学习模型,如集成学习和迁移学习,并分析了这些模型的可解释性方法。作者还着重研究了深度学习方法在分子表示和性质预测方面的挑战和机遇。
一、Introduction
分子性质是许多领域的重要因素,包括化学、药物发现和医疗保健,且与量子力学、物理化学、生物物理、生理学等相关。计算机辅助方法能够快速预测分子性质,在具体实验开始前提供感兴趣分子的总体情况。这些方法被称为定量结构-活性关系(quantitative structure-activity relationship, QSAR)或定量结构-性质关系(quantitative structure-property relationship, QSPR)模型。此外,随着机器学习方法的发展,分子性质预测的准确性和速度也得到了提高,加速了其他相关应用,如药物-靶点亲和力预测和分子合成预测。特别是,作为机器学习的一个重要分支,深度学习方法受到了极大的关注。这种方法可以更精确地发现分子的结构与性质之间的关系。
要研究分子性质的第一个问题是,如何表示一个分子。作者将分子表示方法划分为3类,1维表示、2维表示和3维表示。如图1所示,作者展示了药物imatinib mesylate的三种表示形式。
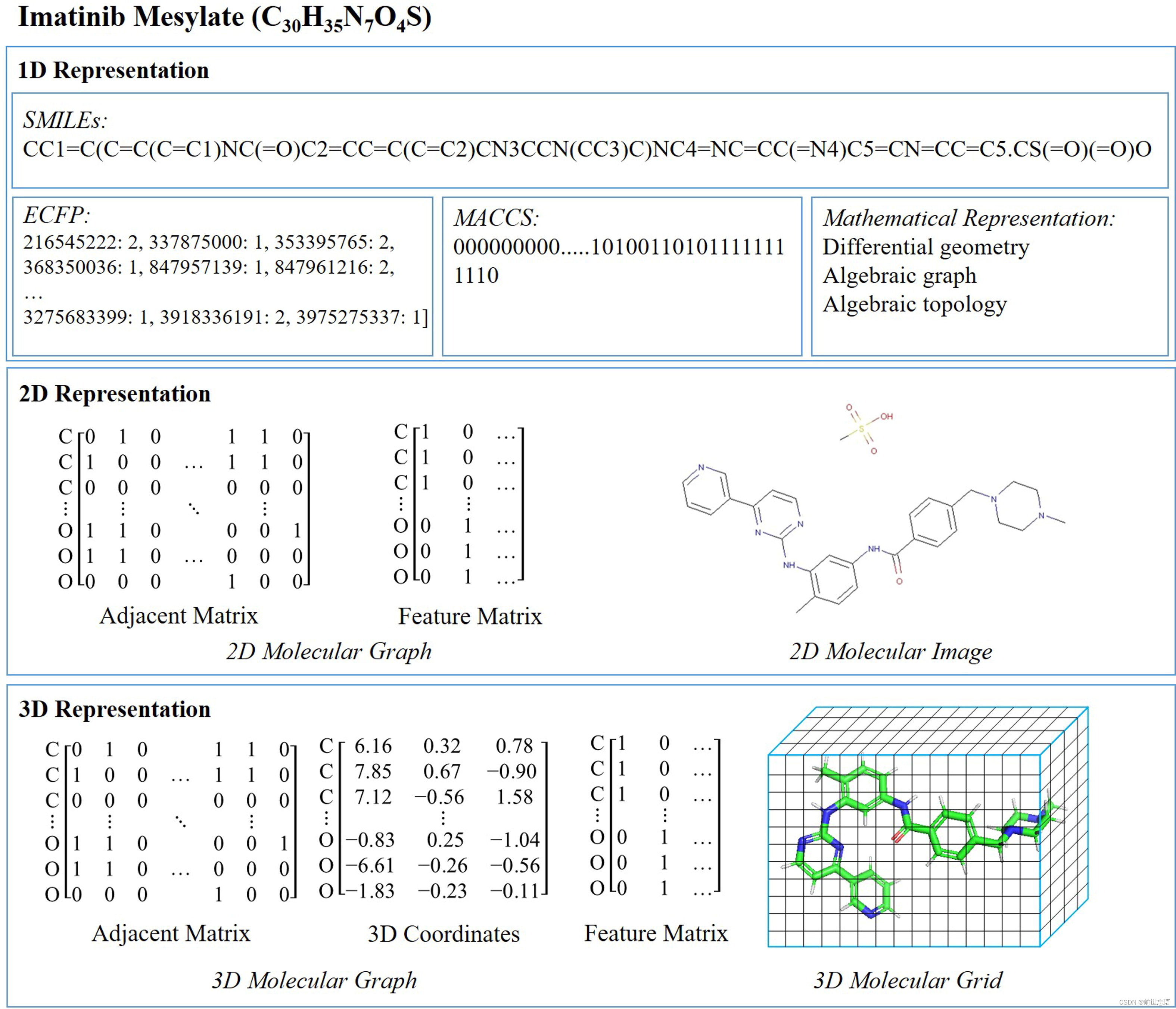
图1 药物甲磺酸伊马替尼(imatinib mesylate,化学式:C30H35N7O4S)的1D、2D和3D表示。(a) 对于1D表示,图中展示了多种表示形式,包括SMILES、extended connectivity fingerprint (ECFP)、molecular access system (MACCS) fingerprint,和一些数学表示方法。(b) 对于2D表示,分子图可以被表示为两个矩阵,即邻接矩阵和特征矩阵。右边的分子图像(由RDKit生成)是另一种2D表示形式。(c) 图中展示了两种3D表示方法:一个3D分子图和一个3D分子网格表示(由PyMOL生成)。
分子式是分子的通用表示(例如,C30H35N7O4S表示甲磺酸伊马替尼)。然而,由于缺乏结构信息,这种表示方式使DL模型难以预测分子的性质。因此,一种更先进的基于序列的表示法,即SMILES被提出,并已成为一种流行的分子表示法。在SMILES字符串中,原子和化学键分别用字母和标点符号表示,分支用括号表示。然而,由于一个SMILES字符串可能不对应一个有效的分子,SELFIES被提出来解决这个问题,其中每个SELFIES字符串对应一个有效的分子。此外,指纹是另一种基于序列的分子表示,它包含了分子结构信息,如ECFP和MACCS(图1)。
分子2D数据中所包含的结构信息对分子2D数据的MPP有很大的帮助,可分为两类:分子图数据(Graph)和分子图像数据(Image)(图1 b)。图数据是学习分子表示的一种有效方法。一个分子的原子被看作是分子图中的节点,而化学键被看作是边。随着图卷积网络(GCNs)的发展,可以更直接和有效地收集相邻节点,这对捕获分子内原子之间的空间关系很有用。与此同时,通过将分子转换为基于像素的分子图像是分子的另一种2D表示格式,图像中的每个像素代表一个键、原子或空白背景。
三维结构提供了关于分子的最详细的信息。与2D分子数据类似,有2种类型的3D分子数据:3D分子图和3D分子网格(图1c)。3D分子图记录了每个原子的3D位置,3D分子网格是一种特殊的3D图像,网格中的体素通过不同的方法表示分子构象的不同元素或属性。
二、Sequence-based methods
SMILES是描述分子最直接、最简单的方法。它类似于自然语言,其中每个原子都是句子中的一个词。鉴于自然语言处理(NLP)领域的快速发展,NLP方法可以应用于SMILES序列的嵌入。作者接着总结了研究者开发的多种基于序列的方法,包括:数据增广方法、卷积神经网络方法、循环神经网络方法、子结构学习方法和基于序列的自监督学习方法等。图2也展示了基于SMILES的两种类型的自监督学习方法。
1.Data augmentation methods
在使用DL模型处理SMILES之前,必须克服其不一致性。对于一个分子,根据SMILES的语法,可能有多个有效的SMILES序列。一个开始的原子和一个遍历顺序可能对应一个序列。因此,可以选择任意一个原子作为起始点,任意一个分支作为第一个经过的原子。规范的SMILES根据一定的规则确保每个分子只有一个SMILES字符串。然而,当非规范的SMILES作为输入时,不同的SMILES格式可以提高DL模型的学习能力。这是因为通过提供与SMILES的语法和化学性质相关的潜在特征,非规范SMILES也可以使DL模型受益。因此,建议数据增强或枚举,以扩大字符串的覆盖率,从而保证模型能够学习一个分子的多个字符串。
假设每个分子的长度不同,那么短序列比长序列有更少的符号。在Conv2S中,SMILES连续生成,直到  ,其中L 和N是生成的SMILES的长度和数量。为了克服数据集不平衡的问题,用重复的SMILES补充含有较少SMILES,以确保所有分子都有相同数目的SMILES。Kimber等人对五种不同的SMILES增强方法进行了综合分析。他们发现,该增强方法提高了DL模型的性能,并且使用规范SMILES得到的结果比使用单一随机SMILES得到的结果好。
2.Convolutional neural network model
卷积神经网络(CNNs)可用于序列数据处理。例如,CovS2S模型将SMILES转换为一个整数列表,然后添加嵌入的位置,以通知模型对应字母的位置。Lim等人还对SMILES进行了字符级的嵌入,其中为每个letter生成嵌入向量。引入一个具有多头自注意模块的CNN层来处理输入嵌入,并增加两个完全连接层来输出预测。SMILES convolution fingerprint (SCFPs)将多个原子的性质(包括类型、度、电荷和手性)组合在一起,形成一个原子的特征向量。SMILES序列可以转换为一个矩阵,该矩阵的长度为SMILES序列的最大长度。构造了两个卷积层和池化层以及全局池化层来提取该表示,并通过回溯,用响应滤波器的最大贡献来表示重要的子结构。
考虑到基于CNN的方法需要固定长度的输入样本,药物SMILES在被发送到网络之前必须被填充或截断。通常,数据集中SMILES的最大长度或平均长度可以为模型输入固定长度的样本。但是,这两种方法都会导致数据丢失和引入噪声,这是基于CNN的方法的主要问题之一。
3.Recurrent neural network models
在处理分子序列数据中,基于SMILES的RNN的准确性和鲁棒性是提取分子特征的关键。Hou等人提出了一种双向LSTM(Bi-LSTM),该方法具有通道和贝叶斯优化的空间注意网络,能够明确识别SMILES中的主要特征。Nazarova等人提出了RNN的两种反向传播方法,并比较了SMILES的二进制和十进制表示在聚合物性能预测中的表现。他们发现二进制表示比十进制表示更准确。CNN和RNN的结合也可以提高表示的性能。Li等人使用one-hot编码将SMILES中的每个字符转换为一个向量,并引入一种堆叠CNN和RNN层的混合体系结构进行表示提取。结果表明,与普通RNN模型相比,CNN能够提高预测性能。
虽然RNN适合于序列处理,但仅使用序列而忽略其他信息(例如化学上下文或分子结构)。因此,仅使用序列并不是学习分子表示的全面方法。特别是,分子内的原子关系、原子基团以及键的类型也可能与分子性质有关,可以通过某种方式引入分子性质来提高性能。此外,基于序列的模型的可解释性还存在缺陷。由于分子分支与主序列融合,如果没有其他更具体的设置,RNN模型很难区分基序和分支。因此,来自相同功能的基团的关键原子可能位于彼此很远的地方。即使引入了注意机制,它也只关注单个字母或相邻字母。
4.Substructure learning methods
官能团是分子的关键部分,分子的性质和活性与官能团和亚结构高度相关。然而,SMILES并不直接包含这类信息。Mol2Context-vec在ECFP的帮助下提取了子结构。该子结构由多个原子组成,包括一个中心原子和所有在中心原子周围给定的半径内的原子。每个子结构都有自己的标识符。子结构序列是Bi-LSTM的输入,它捕获原子之间的相互作用。Mol-BERT将每个子结构的向量总结为无监督学习和下游任务的分子表示。S2DV定义了一个分割字符来保留子结构信息,并定义了一个具有预定义的大小的滑动窗口来处理序列。每个滑动窗口生成一个向量来描述化合物及其子结构之间的关系。
5.Sequence-based self-supervised learning methods
近年来,自监督学习(SSL)得到了迅速发展。SSL可以使用大量的未标记数据集,设计pretext任务来学习数据的内在特征,从而减少对标记样本的需求。基于序列的SSL模型可以分为对比学习方法和生成学习方法。对比学习方法是通过构造伪标签数据来学习正样本和负样本之间的差异,而生成学习方法则是将输入编码为潜在特征,然后解码重建输入,从而将潜在特征作为输入的表示。对比学习方法从数据信息的角度寻找数据间信息,而生成学习方法关注数据内部信息。
在NLP领域,来自Transformer(BERT)的双向编码器表示是一种广泛使用的SSL方法,其中一个Transformer包括一个编码器和一个解码器。BERT-like可以应用于SMILES序列中提取原子或分子特征(图2)。MOL-BERT结合三个任务生成分子表示。第一种是BERT中使用的MLM。第二种是SMILES等效法,将个相同分子的两个SMILES序列作为一类,将个不同分子的两个SMILES序列作为另一类进行训练。第三种是利用分子化学特性进行预测。所有三个模型被联合训练输出分子表征。SMILES-BERT也是基于BERT的,但只保留了MLM,并引入了一个自注意层来使用顺序信息。
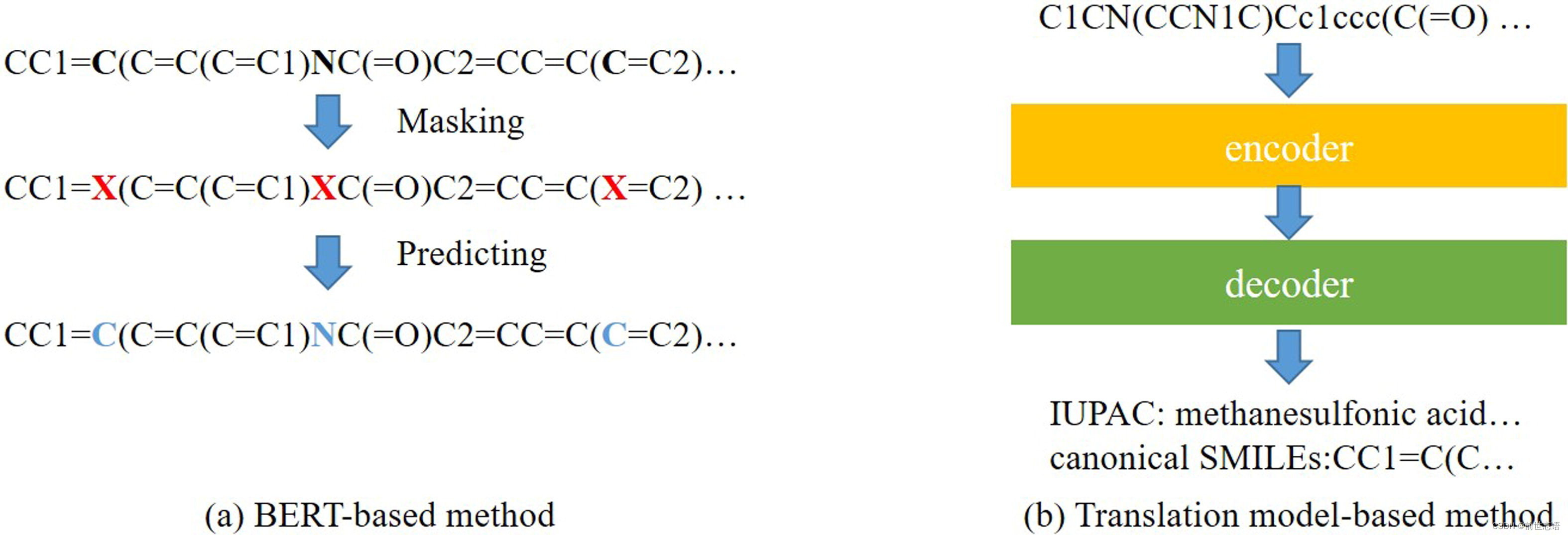
图2 基于SMILES的不同类型的自监督学习(self-supervised learning)方法。(a) 基于transformer(BERT)的双向编码表示。SMILES序列被用作输入,并随机mask一些原子。然后训练语言模型来预测这些被mask的字符,进行表示学习。(b) 基于翻译模型的方法。该模型将输入的SMILES序列变换为另一种类型的序列。编码器编码的隐特征被用作分子表示
除了基于BERT的方法,还有其他生成式方法使用编码器-解码器架构来表示分子。Hu等人使用基于GRU的编码器-解码器生成模型生成固定大小的潜在特征来从SMILES中学习分子,并引入CNN模型进行下游预测任务。NLP中使用的翻译模型也可以在SMILES中实现(图2b)。序列-序列(seq2seq)是一种流行的工具,包含用于翻译任务的编码器和解码器,其目标是将一个序列转换为另一个序列。同样,编码器的输出也可以是其他任务的表示。当对SMILES使用seq2seq方法时,主要目标是找到两个相对应序列进行训练。Winter等人提出了一种方法,用于SMILES和IUPAC名称之间的翻译。对于基于序列的SSL方法,对比学习方法能够帮助发现SMILES之间的相互关系,这是分子表征的未来研究方向。
三、Graph-based methods
图是一种更直接的结构,可以存储和表示大多数结构信息。在图模型中,原子被表示为节点,化学键被表示为边,每个节点都有自己的特征。在图数据的帮助下,分子内的结构信息可使用GCNs提取。GCNs能够捕获有关连接节点之间关系的信息。一般来说,GCN有两种类型:空域卷积和频谱卷积。前者通过在空域中使用特定的消息传递规则收集相邻节点的信息来更新每个节点的特征。后者通过对拉普拉斯矩阵进行特征值分解,将图数据转换为谱域提取特征。接着,作者回顾了领域内的相关方法,包括谱域GCN模型、空域GCN模型、基于树的方法和基于图的自监督学习方法等。图3展示了基于图的三种不同类型的自监督学习方法。
1.Spectral GCN models
LanczosNet使用Lanczos算法构建了用于谱域GCN的低秩拉普拉斯近似,用于利用多尺度信息。Shang等人从Chebyshev近似出发,提出了一种一致的边缘感知多视角谱域GCN,该模型采用了一种新的flexble谱域滤波器,根据边的类型将分子图分解为图的多个视图,并采用学习边的注意权重的一致边映射方法来保证边一致性。因此,在分子表征和性质预测方面,谱域方法比空间方法少。这是因为空域GCN模型能够将不同原子的分子生成不同大小的图,而谱域GCN模型只能处理固定大小的图。因此,在处理输入图数据样本时,仍然需要进行数据对齐操作,如填充或截断,这将损害数据的完整性,并影响模型的最终性能。
2.Spatial GCN models
空域GCN模型在药物发现和MPP中应用更为广泛。一般来说,空域GCN模型需要两个矩阵作为输入:一个邻接矩阵和一个特征矩阵。前者表示原子之间的空间联系,可以从分子图中得到。DeepAtomicCharge使用带有跳连接的消息传递神经网络(MPNN)来预测原子电荷。AttentiveFP是由GCN派生而来的另一种分子表示方法,它自动学习非局域分子内相互作用,并通过注意机制捕获指定任务的隐藏边缘。多物理GNN结合了尺度特定的图神经网络和元素特定的图神经网络,从不同的尺度捕获各种原子相互作用,以实现多物理表示。
边也被认为是卷积过程中应该考虑的一个重要因素。交叉依赖的图神经网络认为原子和化学键同样重要。原子中心视图和键中心视图都被构造起来了,并且在这两个视图之间提出了一个交叉依赖的消息传递方案。TrimNet提出了一种三元注意边缘网络,通过原子-边-原子排列来收集信息,提高边缘信息的提取。一对原子和它们之间的键被连接成一个三元组,多头注意被用来从相邻的节点和边缘收集一个节点的信息。
3.Tree-based methods
通过选择一个起始原子,可以将图转换为树形结构。此外,根据广度优先遍历(BFS)或深度优先遍历(DFS)方法,利用RNN模型将树转换为原子序列进行表征学习。Su和Wang等人都开发了基于分子树结构的QSAR建模方法。将分子编码为特征描述符,利用树状LSTM(Tree-LSTM)对分子树结构进行描述,并将其与分子性质关联起来。Junction Tree首先将分子分解为子结构,然后根据这个子结构生成树状图。虽然Junction Tree被提出用于分子生成,但编码器部分可以被分离出来进行性质预测。受此启发,Wang等人提出了一种基于多通道树的分子预测方法。将分子转换为基于子结构的图,并应用BFS方法遍历图,生成树形结构。在此基础上,建立了一种基于GRU的具有注意机制的神经网络,对分子特征进行了多层次的学习。
然而,由于不同的遍历方法,如BFS或DFS,以及不同的根原子选择方法,树的结构不是唯一的。生成的分子树的多结构会影响模型的泛化。规范结构的定义并不健壮,不能确保所有与属性相关的信息,特别是连接信息在结构中得到全面的显示。因为将一个图转换到树将不得不分解一个或多个连接。
4.Graph-based self-supervised learning methods
与序列相似,SSL方法在图数据上也取得了显著的性能。Wu等人在图数据上定义了另一种类型的SSL方法,预测方法,该方法基于自生成标签的prediction-based pretext任务。
在对比学习方法中,如何构建正样本和负样本是至关重要的一步。在这方面,MolCLR通过三种方式增强分子:原子掩蔽、键删除和子图删除。来自同一分子的样品记为正例,其他的记为负例,用于对比损失计算。然而,MOCL认为增强方法,如删除节点,边扰动,子图提取,会影响分子的性质,不适合对比学习。取而代之的是,MOCL采用了一种子结构替代策略,其中一个子结构被具有相似属性的bioisostere所取代。在NLP中,许多特征提取在单词和句子层次上。NLP中的这两个层类似于图中的节点层和图层,它们分别代表局部特征和全局特征。Li 等人在节点和图级别对模型进行了预训练。在图的层次上,每个分子被分解成两个部分,模型需要预测这两个部分是否来自同一个分子。分子图上对比学习方法的过程如图3a所示。
生成方法通过编码器-解码器模型重构输入。分子图BERT将局部消息传递和GNN结合到BERT模型中,预测指定掩码原子的类型,用于模型预训练。Koge等人使用分子超图语法VAE提取分子的嵌入,将分子的结构和物理性质嵌入到VAE的潜在特征中。基于分子图的生成学习方法的过程如图3b所示。
通常在MPP中使用的预测模型用于预测节点或边的类型或属性。GROVE组合了两个SSL。第一个SSL任务(在节点/边级别定义)是预测子图的属性。第二个SSL任务(在图级别定义),是预测不同的基序的出现。结合这两个层次可以提供分子的结构和语义信息。此外,SSL和监督学习的相互关联为理解分子提供了一种新的途径。例如,SUGAR结合了基于子图的监督方法和SSL方法,来自分类和互信息最大化的两个损失被分组到最终损失函数中。评价结果表明,引入SSL后,模型的性能得到了改善。基于分子图的预测学习方法的过程如图3c所示。
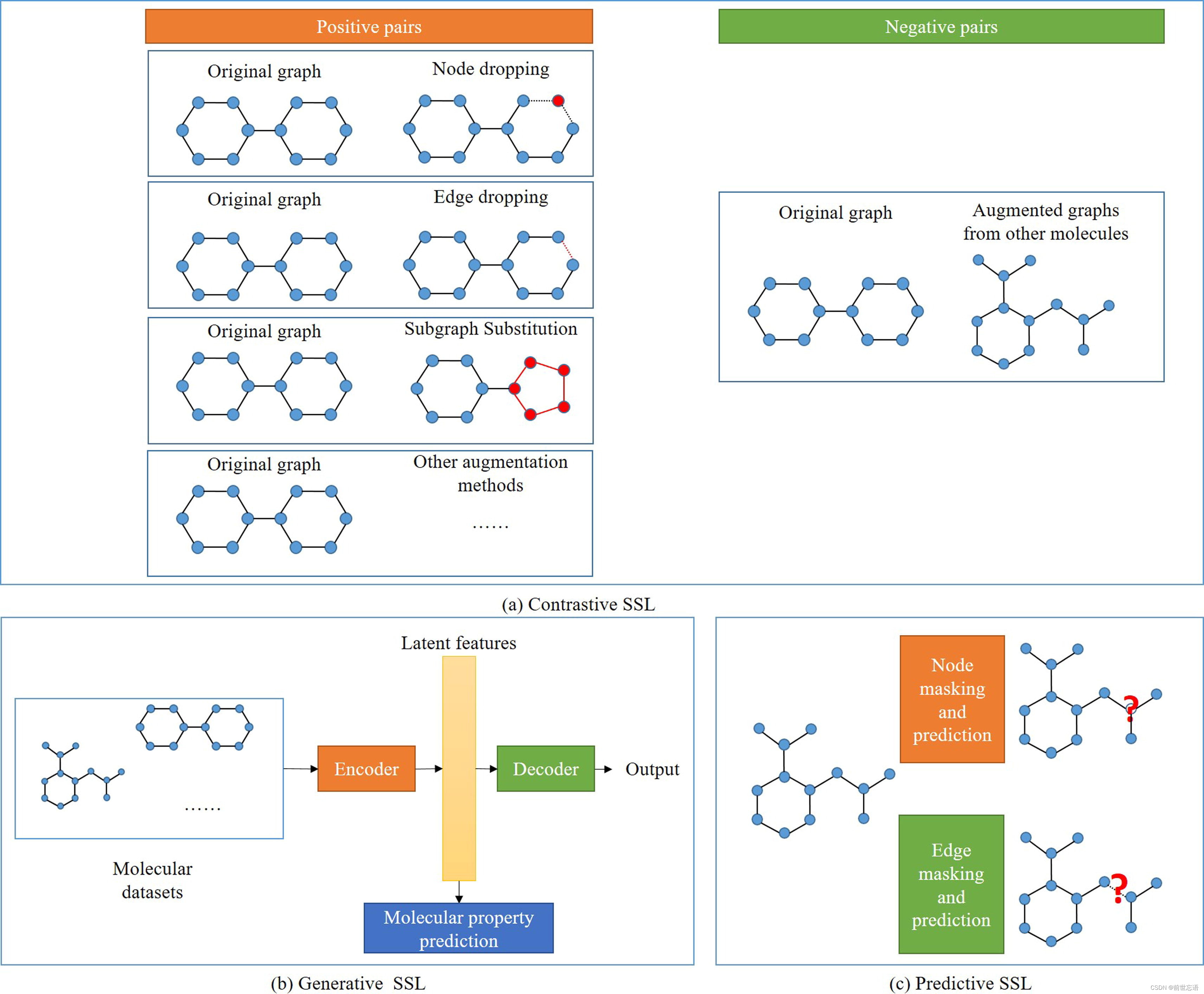
图3 使用图的不同类型的自监督学习方法。(a) 对比自监督L:使用数据增强方法,如对正样本对的节点drop、边drop和子图替换,而对负样本对随机选择其他增强图。(b) 生成自监督:通过编码器-解码器模型重构输入,以获取用于表示的隐特征。(c) 预测自监督:随机mask一些节点或边,让模型预测结果;通过这样做,该模型能够学习到隐特征并生成有效的分子表示。
四、Image-based methods
DL方法在图像处理领域取得了巨大成功,这也为QSAR/QSPR的研究奠定了基础。更具体地说,分子可以转换成图像,因此传统的深度学习模型可以用于QSAR/QSPR任务,其中CNN是最广泛用于分子特征提取的。
对于基于图像生成的方法,最简单的方法是直接使用分子图像,其可以通过RDKit和Open Babel等软件进行转换。然而,这种类型的图像引入了一个很大的空白区域,没有有效的信息。此外,尺度敏感性是另一个问题,因为所有分子都被转换成相同大小的图像。由于整个分子图像的固定大小,相同原子/结构的大小在不同的分子中是振动的。其他的图像生成方法试图避免这个问题。Yoshimori等人为每个原子生成map,并将所有原子map加在一起形成molecular topographic map作为28*28热图。MolMap将分子描述符和指纹特征映射到二维特征映射中,从而形成一种方法,将手工制作的特征组合到二维空间中,以捕获分子特征的内在相关性。
基于图像的方法不是MPP的主流方法,因为它们必须将数据样本转换到欧氏空间,由于缺乏原子和键属性,不适合分子性质预测。为了利用基于图像的DL模型,能够从某些角度发现原子间关系的图像生成方法值得进一步研究,这将有助于生成新的有效的分子表示。
五、3D Graph-based methods
分子的构象通常包含分子的原子3D坐标,也称为几何数据,可以为分子性质预测提供额外的空间信息。首先要解决的问题是三维分子晶体数据集有限。为了扩大基于几何的方法的应用领域,可以基于二维数据生成构象。RDKit中的MMFF94可用于构象生成。此外,哈密顿神经网络也可以用来预测分子构象,并将预测的三维坐标输入到基于MPNN的分子指纹生成器中用于分子表示。
基于3D图形的方法主要是通过引入几何信息对GCN进行调整或改进。除了邻接矩阵和特征矩阵,3D-GCN引入了一个由原子间3D位置组成的相对位置矩阵,以确保卷积过程中的平移不变性。结果表明,训练后的模型具有旋转随机性的内在特性,无需额外的增强方法。Lu等人设计了一种从分子的构象和空间信息中捕获多能级量子相互作用的GCN。该模型将相邻节点的不同阶数依次包含在节点中,以保证节点表示能够覆盖高阶相互作用。然而,不同于其他大型图数据,如社交媒体和引文网络图,分子图的节点数量是有限的。在某些情况下,高阶邻域可能会覆盖一个分子中的原子,从而导致over-smoothing。
为了解决三维图中的消息传递问题,提出了球形消息传递。具体来说,每个原子的3D坐标被转换为球面坐标系,原点节点的邻近节点由一个由边长、边间角度和扭转角组成的三元组指定。与普通的3D坐标相比,三元组更加灵活,能够更准确地描述分子的结构。球形信息传递对输入分子的平移和旋转是不变的。此外,几何消息传递神经网络(GemNet)还使用了分子的球面表示,以确保模型对平移不变,对置换和旋转等变。
此外,SSL方法是基于3D图的方法的一个重要分支,用于发现图的不同特征。Fang等人提出了一种充分利用分子几何信息的自监督框架。他们构建了一个新的键角图,其中分子中的化学键被视为节点而不是边,而两个键之间形成的角被视为键之间的边。对某些原子的跳邻域项进行掩码,预测键长和键角,提取局部表示。Liu等人提出了一种SSL方法,包含分子的3D和2D视图之间的对比学习和生成学习方法。在对比学习中,将来自同一分子的三维和二维图形作为个正对来训练模型。在生成式学习中,模型被训练成从二维拓扑生成三维构象。结合两种策略进行分子表达。
六、3D Grid-based methods
3D网格是另一种使用分子几何数据的表示方法,它将每个原子放置在一个或多个网格中。事实上,蛋白质等大分子可以用3D网格更好地表示,在MPP中仍然表现出良好的性能,特别是在某些量子力学特性方面。3D CNN是3D网格数据的最佳选择;因此,为3D CNN创建一个功能更强大、信息更丰富的网格可以提高分子性质预测的性能。
Libmolgrid提供了一个生成3D分子数据体素网格的库来表示分子。网格的分辨率是影响结果的一个重要因素。多分辨率3D-DenseNet采用原子中心高斯密度创建3D网格,生成不同尺度的多通道网格(4-14 Å)作为3D-DenseNet处理和预测的输入数据。Casey等人还使用高斯模型通过生成了两个三维空间点网格(电子电荷密度和静电势)。在上述两种方法中,也应用旋转增强方法对序列数据集进行放大,而不是改变其大小。Tran等人选择C, H, O, N, S, Cl作为3D网格的独立通道,并使用基于CNN的自编码器进行分子表示。kuzminykh等人发现分子的常规三维网格过于稀疏,影响了CNN的性能,于是提出了一种波变换平滑方法来填充每个原子的空洞体素。
但是,基于3D图形和基于3D网格的MPP方法仍然缺乏。分析时间长是一个严重的问题,尤其是基于3D网格的方法。此外,3D图数据上的图卷积仍然是一个悬而未决的问题。通过简单地添加3D位置信息将传统的GCN方法扩展到3D场景,并不能充分利用几何数据的优势。虽然球形消息传递试图将特定的GCN贴合到3D图上,但设计3D图消息传递机制的一种有前途的方法。
七、Hybrid data-based methods and ensemble learning
所有上述的1D、2D和3D表示方法都以不同的方式表示分子,将它们结合起来可以提供一个分子的多视图。GraSeq结合分子图和SMILES序列,使用GCN和biLSTM进行编码。Karim等人将SMILES、指纹、分子图、2D和3D描述符与多个DL模型相结合,用于定量毒性预测。指纹通常被认为是一种常用的辅助因子,基于图像的和基于图的方法都可以将指纹结合起来以提高预测性能。
集成学习也可以连接多个分类器,以提高联合模型每个单独分类器的性能。Kosasih等人建立了三个GINs的集合。Busk等人还构建了多个MPNNs的集合,用随机参数对其进行初始化,并对其进行单独训练。Karim等人将SMILES字符串转换为一个one-hot向量,表示每个字符缺少的字符,将分子图像和2D数字特征作为输入,并分别对这三种类型的数据训练RNN、CNN和FCN。这些网络的输出通过集成平均法或元神经网络进行组合。
八、Transfer learning, multi-task learning, and meta-learning
数据集的不足是MPP中的另一个问题。使用来自其他域的大型数据集来帮助找到数据较少的目标域的模式将有效地克服这个问题。为此,建议使用迁移学习、多任务学习和元学习。
对于迁移学习,模型首先在大数据集上进行训练,以完成某些pretext任务,从而学习分子的一般表示。然后将所学的一般表示法用于下游任务(通常具有有限的样本),以传递先验知识。MRlogP在大数据集(500000)上对低准确度的预测logP值进行传递学习,并在244个药物类化合物的小准确度数据集上对参数进行微调。Zhong 等人在ImageNet上使用预训练,并将其传输到用于QSAR任务的分子图像数据。
多任务学习同时训练所有任务,并共享表示,以提高预测的通用性。Liu等人使用多任务学习从QM9和Alchemy数据集预测12个量子化学性质。此外,在框架中选择了一个以原子为中心的对称函数作为辅助预测目标,以提高泛化性和可迁移性。
近年来,出现了元学习方法来解决少数问题,也称为“learning to learn”。在训练过程中,元学习将训练数据划分为不同的元任务,以学习初始化良好的模型参数,并具有较高的泛化能力。模型在新任务上通过少量梯度下降进行更新,以提高模型的性能。例如,Meta-MGNN将图形神经网络、SSL和任务中Tox21的权重感知元学习与MPP的SIDER中的六个任务相结合。Wang等人提出了一种属性感知嵌入方法,该方法考虑了不同分子属性和不同分子亚结构之间的关系,并且元学习方法选择性地更新任务中的参数,以分别对泛型和属性感知知识建模。因此,所有三种类型的学习方法都可以找到不同任务之间的关系,并解决数据有限的问题。
通常,迁移学习方法可以使用大规模数据集,如ChEMBL和ZINC,来学习分子的一般表示,并对其进行微调以适应特定的数据集。多任务学习通过同时学习几个相关任务来改进模型的泛化能力,而元学习用有限的数据预测看不见的任务。
九、Interpretability of the DL model on molecular properties
DL最有争议的领域是其可解释性。可解释DL方法分为两类:被动和主动。被动方法使用DL模型中的参数进行解释,而主动方法改变训练过程以提高模型的可解释性。
Jiménez Luna 等人指出,透明度、正当性、信息性、和不确定性估计是药物设计中人工智能方法的可解释性的主要方面。Pope等人在GCN上评估了三种显著的可解释性方法,称显著子图可以作为官能团被解释。Jiménez Luna等人提出了一种特征属性方法,使用经过训练的MPNN模型为每个节点生成重要性分数,并根据原子的重要性分数将其着色。该方法能识别700个药效团基序,并能识别属性。对于分子性质预测,被动方法仍然是了解分子精确亚结构与其性质之间关系的主要方法。
注意机制可以学习输入的7不同部分的权重,以确保DL模型能够专注于重要部分。注意机制的概念也可用于DL模型的可解释性,其中可以找到重要的原子或基团及其对分子性质的相应贡献。例如,Tang 等人可视化了自注意值,以检测分子的哪些部分有助于其亲脂性或水溶性。Wu等人为毒性预测开发了一个多任务图注意力框架。该框架提取了分子亚结构的特征,每个亚结构被赋予一个模式权重。
此外,不确定性估计是评估模型可靠性的重要方法。Ryu等人使用贝叶斯GCN预测分子性质。它将721标准dropout替换为“Concrete”dropout,并通过贝叶斯方法估计了模型的不确定性。作者发现,不确定性可以作为预测的置信指标。Hirschfel等人对分子性质预测的不确定性估计进行了评估,并提出将弱不确定性估计值结合起来,可以获得更加一致的性能。
十、Molecular property prediction challenges and future work
1.Self-supervised learning methods in 3D
SSL方法是发现分子显著特征的一个有希望的方向。对于SSL方法,设计pretext任务是最关键的一步。然而,隐藏在三维分子数据中的丰富信息仍有待充分利用。3D的SSL方法已经应用于许多领域;因此,设计一种新的分子3D SSL方法将对预测分子性质非常有用。此外,虚拟筛选用于寻找具有高结合亲和力的配体-靶对,这是药物发现和设计的关键步骤。在虚拟筛选软件中,需要目标配体和配体的构象。因此,精确和全面的分子3D表示也将有助于虚拟筛选。
2.Graph convolution methods with experience
GCN方法因其在图数据上的优异性能而成为分子相关任务中的主流方法。然而,仍有改进的余地,如引入经验数据和专家知识等。我们不能忽视人类经验对DL模型的影响,通过定义原子、键和官能团的类型,有一些向模型注入专业知识的方法。然而,一些高级或复杂的领域经验在MPP中可能更为强大,例如基序和属性之间的关系,这些尚未被充分利用。
3.1D, 2D, and 3D data fusion and selection methods
通常,与低维数据相比,高维数据包含更多信息。如果是这样的话,那么基于2D数据的模型的性能应该比基于1D数据的更好。然而,正如我们上文所讨论的,混合方法使用1D、2D和3D数据,消融实验已经证明了它们在MPP中的各自作用,表明低维数据提供的信息没有完全被高维数据覆盖。这里提出的一个问题是,当高维数据(2D或3D数据)已经使用时,为什么1D数据仍然有助于模型。这可以从以下两个人的观点中得到回答。首先,将一维序列转换为二维图形时,会丢失一些信息。第二,DL模型不能充分利用高维数据中的隐藏信息。因此,1D数据仍然用作辅助信息源。无论什么原因,如何确定合适的数据类型(或多种类型的最佳组合)仍然是一个悬而未决的问题。
4.Meta-learning methods
迁移学习、多任务学习和元学习都被用于解决某些属性的实验数据不足的问题。作者认为,元学习方法是目前最有前途的研究方向之一。更具体地说,对于实际应用来说,元学习是一种理想的方法,因为当传统的ML或DL模型由于数据样本数量有限而无法使用时,某些任务可能只有少量实例(例如,预测一些罕见的分子性质)。因此,MPP的元学习方法值得进一步研究。
5.The interpretability of DL models
与图像处理中的传统任务不同,大多数分子相关任务都是高度专业化的,需要化学专家来分析潜在机制,例如分子亚结构的作用。分子相关任务的这些特征与DL模型的“黑箱”性质有些矛盾。因此,改进DL模型的解释始终是必要的。更具体地说,通过分析成功预测的和失败的数据样本,在模型中定位关键功能元素,不仅有利于DL模型的最终性能,而且有利于发现新的QSAR理论。作者认为,主动方法是更强大的工具,通过向DL模型添加特定参数来提高其可解释性。
参考(具体细节见原文)
原文链接:Redirectinghttps://doi.org/10.1016/j.drudis.2022.103373
Drug Discov Today | 分子表示与性质预测中的深度学习方法在论文中,作者回顾并总结了现有的分子表示与性质预测的深度学习方法,并讨论了深度学习方法在分子表示和性质预测方面的挑战和机遇。https://mp.weixin.qq.com/s/5ZnW-GF-Jmp3cgrmfXr4Hw
版权归原作者 前世忘语 所有, 如有侵权,请联系我们删除。