
基于构效关系模型的药物设计(QSAR)
定量构效关系(QSAR,Quantitative Structure-Activity Relationship)分析是指利用理论计算和统计分析工具来研究系列化合物结构(包括二维分子结构、三维分子结构和电子结构)与其生物效应(如药物的活性、毒性、药效学性质、药代动力学参数和生物利用度等)之间的定量关系。
它是药物研究中的一个重要理论计算方法和常用手段。例如:
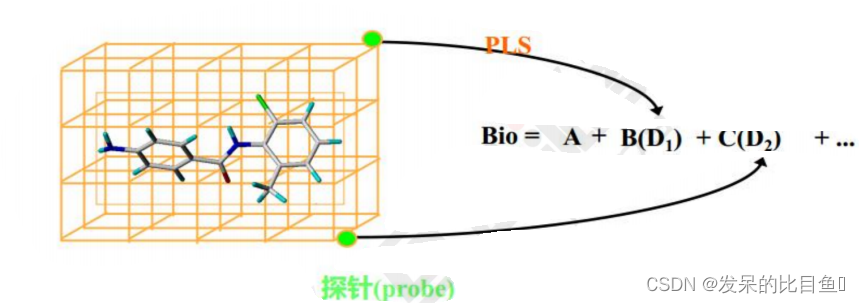
定量构效关系QSAR—揭示一组化合物的生物活性与其分子结构特征之间的相互关系,以数学模型表达和概括出量变规律,以此设计新的化合物
活性
=
f
(分子或片断性质)
活性=f(分子或片断性质)
活性=f(分子或片断性质)
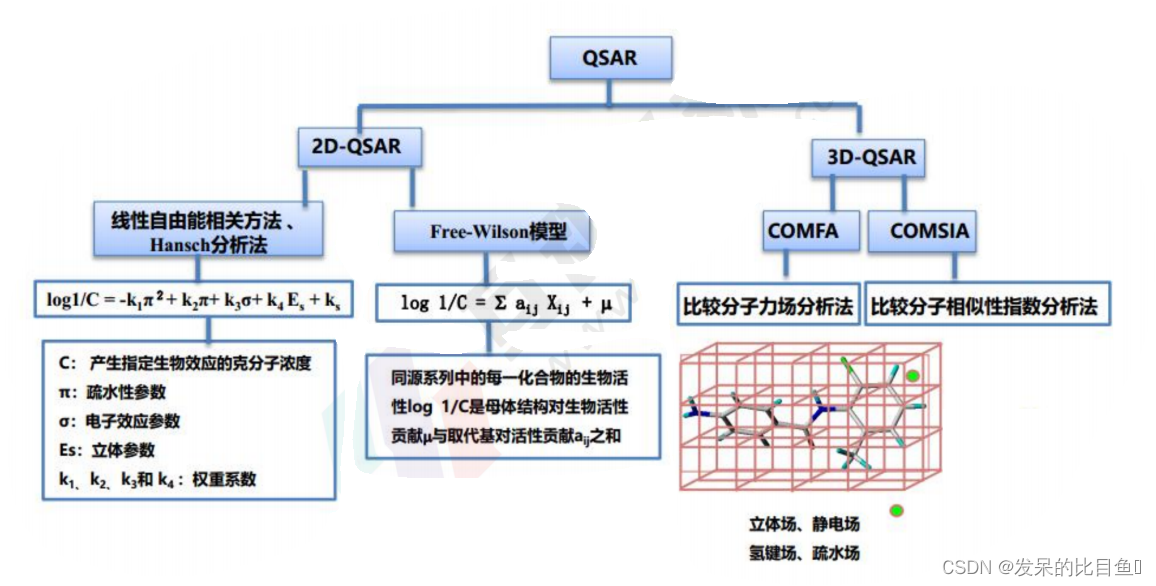
3D-QSAR的基本流程
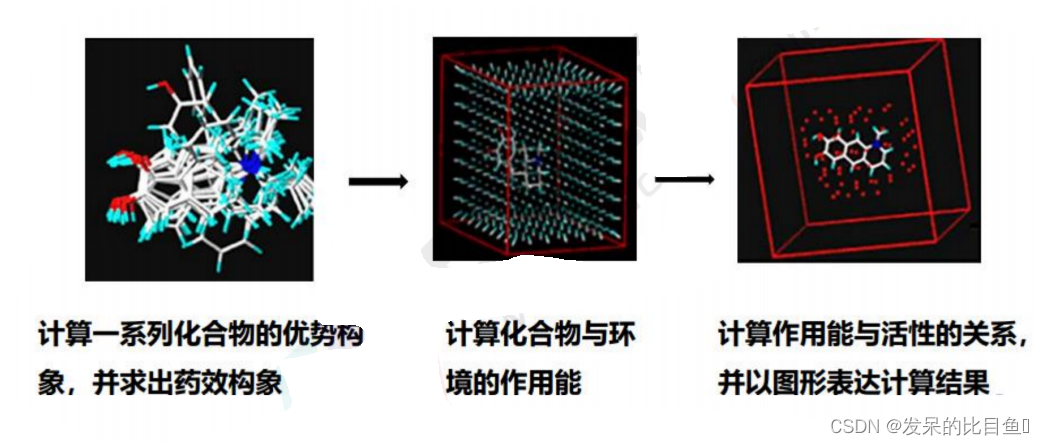
COMFA(Comparative Molecular Field Analysis):比较分子场分析
基本假设:
药物与受体之间只有非键相互作用,没有形成共价键药物活性与立体场和(或)静电场的改变相关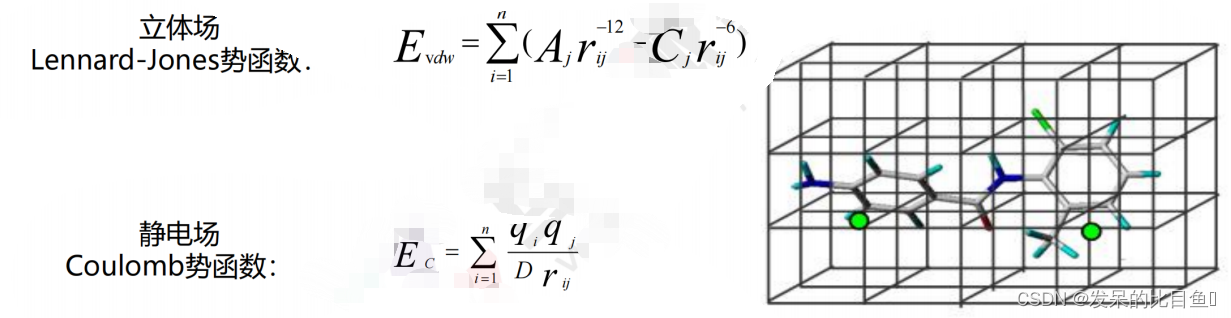
COMSIA(Comparative Molecular Similarity Indices Analysis):比较分子相似因子分析法
作为COMFA的拓展:
除立体场和静电场外,还考虑氢键场和疏水场,这两种作用对药物活性有重要影响改变分子场能函数,克服COMFA计算分子场时在某些格点出现显著变化的缺点由于力场考虑更全面,三维构效模型更优。
A
F
,
k
q
(
j
)
=
∑
i
w
probe,k
w
i
k
e
−
α
r
i
q
2
A_{F, k}^q(j)=\sum_i w_{\text {probe,k }} w_{i k} e^{-\alpha r_{i q}^2}
AF,kq(j)=i∑wprobe,k wike−αriq2

基于构效关系模型的药物设计(QSAR)
3D-QSAR的优缺点
√不必知道靶点的结构 ×预测仅限于由训练集包络的空间之内
√不必输入实验测定或理论计算的理化参 ×不能可靠地预测出原模型范围之外的数值取代基结构
√给出可视图易于解释QSAR结果 ×分析的准确性取决于采用的空间结构
√不限于研究相似分子结构,只须有相同的药效团以相似的方式与靶点作用
√可预测新分子的活性,而不必先合成
COMFA/COMSIA流程

以Imatinib类似物为例构建COMFA与COMSIA模型
数据集基本要求及基本处理
口 化合物的活性值应跨越2-3个数量级
口 化合物在不同数量级上分布均匀
口 训练集分子数目最少为10个,一般15个或以上
口 活性值转化为对数值,一般使用pIC50
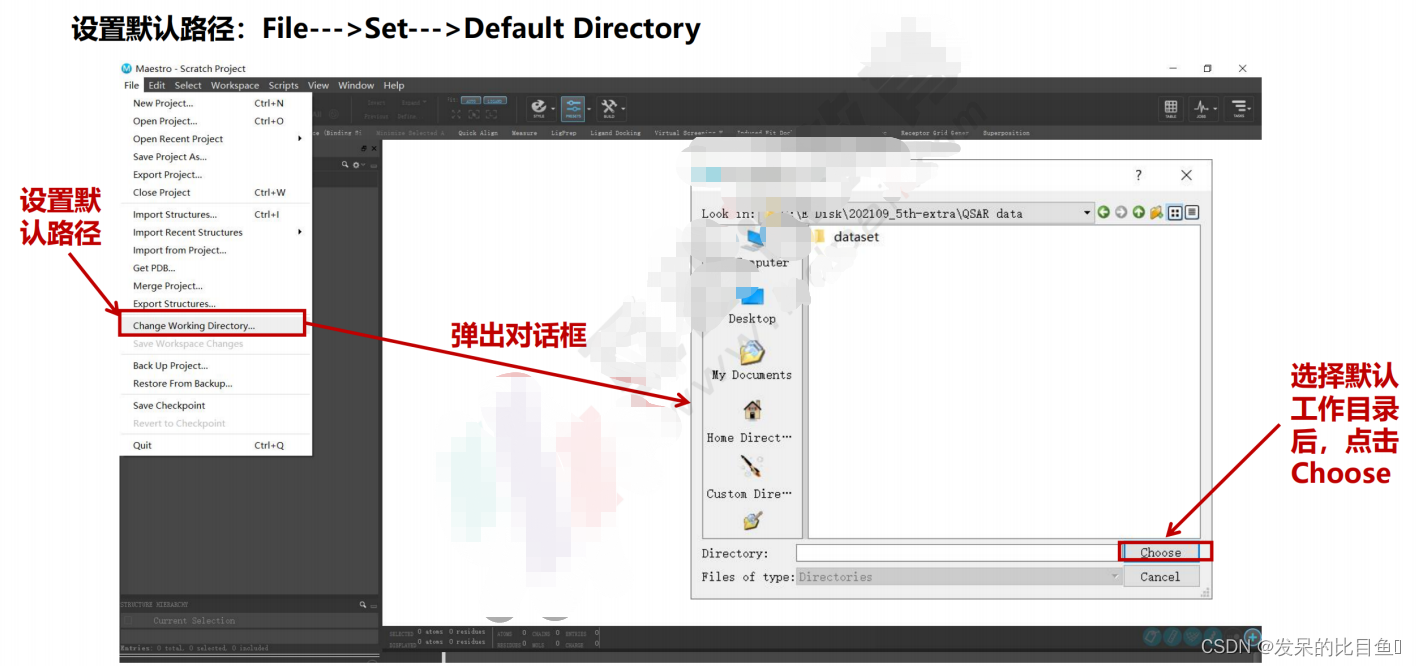
设置默认路径:
File--->Set--->Default Directory
分子准备
数据集优化:
Tasks--->LigPrep
模块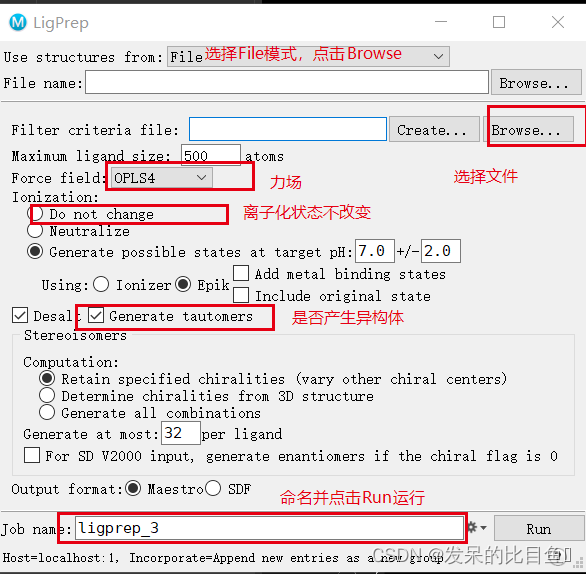
点击左侧分子目录,可选中优化后的分子,并显示在窗口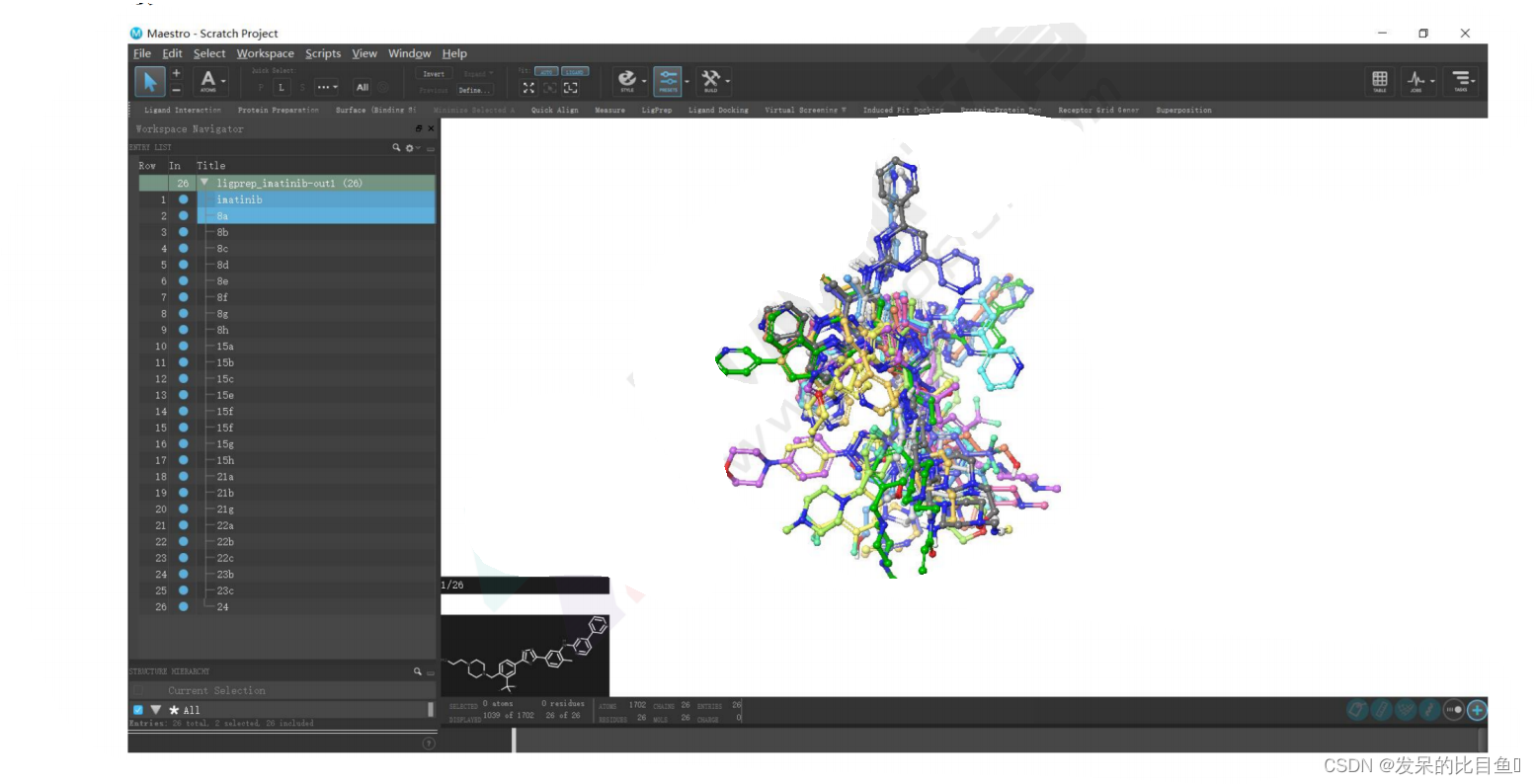
分子叠合:点击
Tasks--->Structure Alignment--->Ligand Alignment
,打开
Ligand Alignment
对话框

分子叠合方法概述:
Ligand Alignment
,指基于柔性形状的分子叠合该方法用
ConfGen
进行配体构象搜索。之后由
ConfGen
生成的构象并选择与参考配体重叠最好的构象,依次与参考配体对齐;
Common scaffold alignment(requires Phase license)
,指基于公共骨架的叠合,有三种模式:
①
Largest common Bemis-Murcko scaffold
使用包含完整环和连接环的1inkers的最大的公共骨架。勾选Use fuzzy matching(使用模糊匹配),表示将所有的非氢原子等同对待,但要区分键的类型(单键、双键、三键、芳香键)。
②
Maximum common substructure
使用所有配体中最大的公共子结构。这比
Bemis-Murcko scaffold
更普遍,因为它不限于环和
linkers
。
③
SMARTS
使用指定的SMARTS模式。可以将SMARTS模式输入或粘贴到文本框中,或者通过单击
Get from selection
从工作区原子选择中生成SMARTS模式。即我们可以自定义公共骨架进行叠合;如我们在工作区中通过按住Ctrl选择我们自定义的公、共骨架,然后点击
Get from Selection
按钮即可;
3D-QSAR模型构建:
step 1. 分子导入
点击
Tasks
—>输入
qsar
–>选择
3D Field-Based
,打开
Field-Based QSAR
面板
分子导入

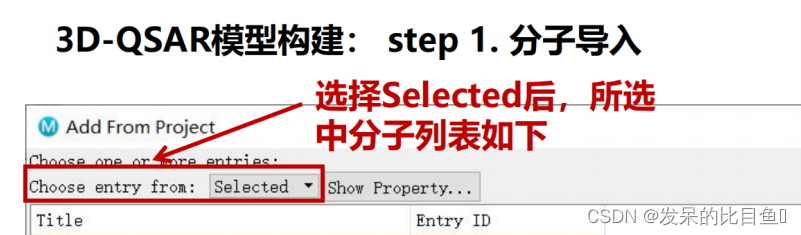

导入分子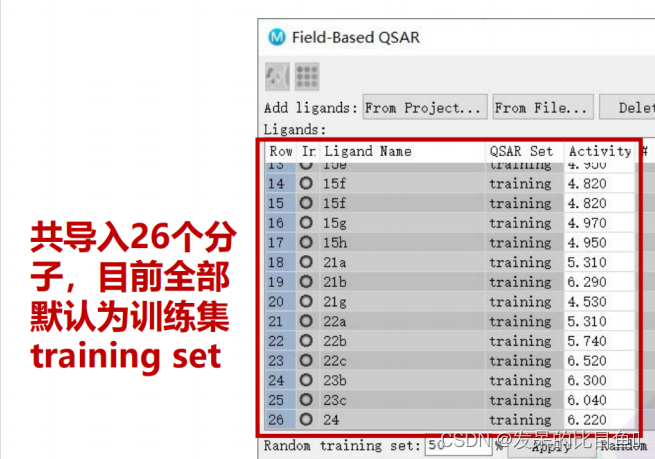
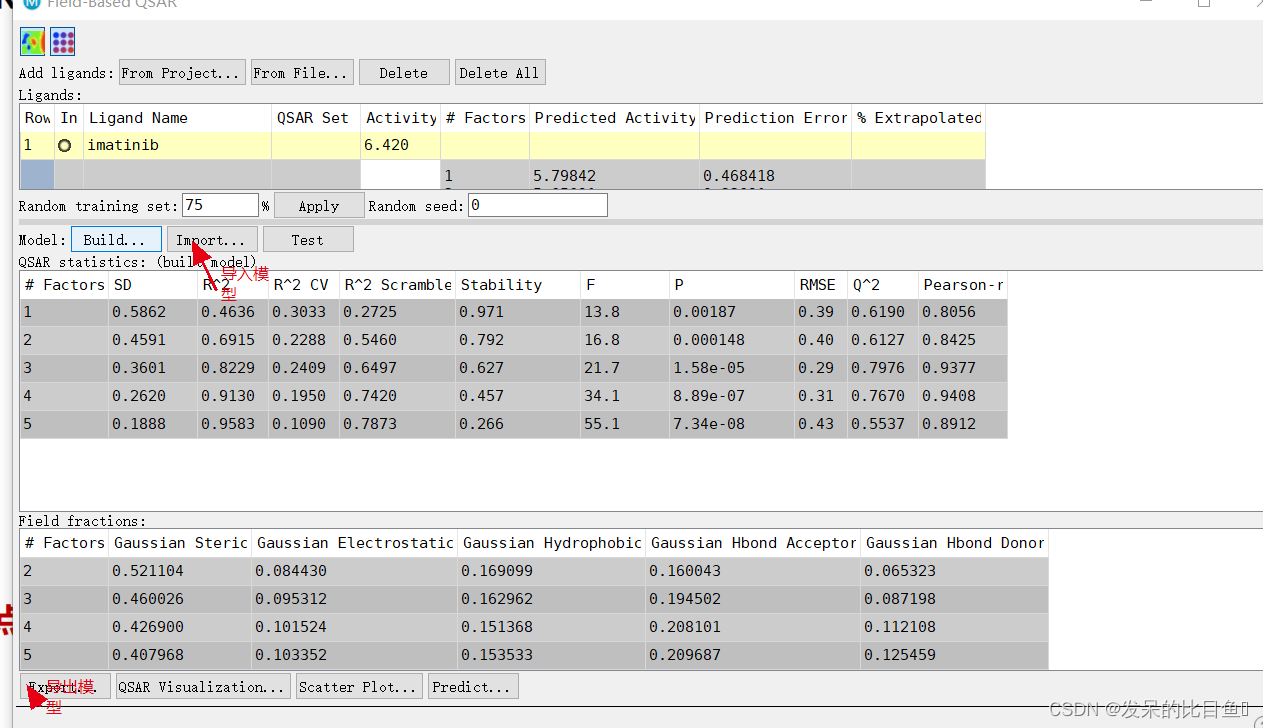
构建训练集和测试集
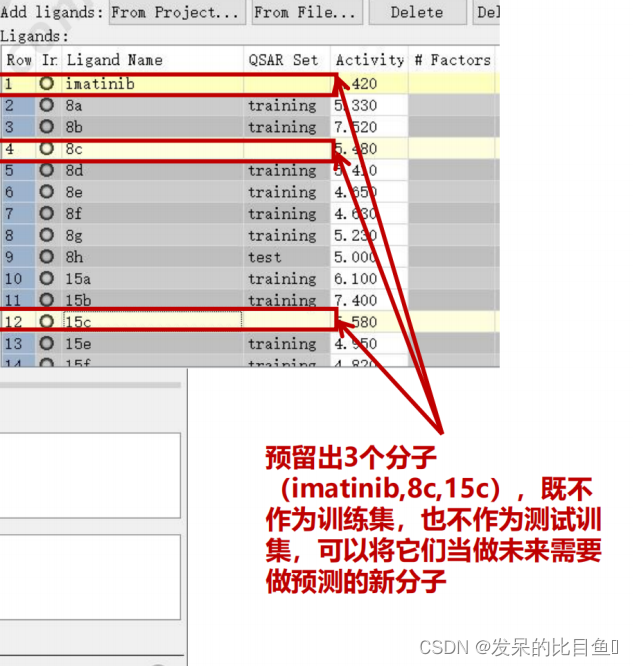
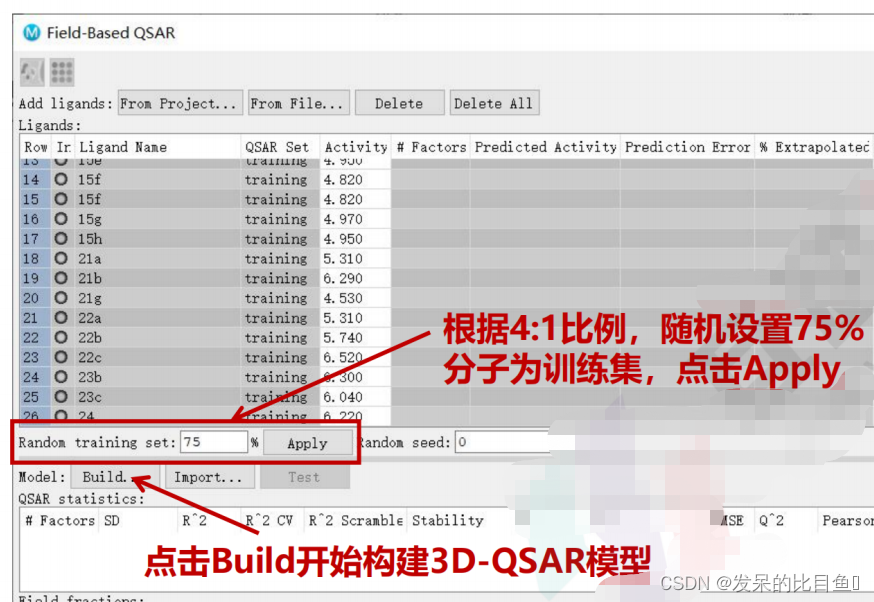
点击Build后,出现Build Field-Based Model

模型结果分析
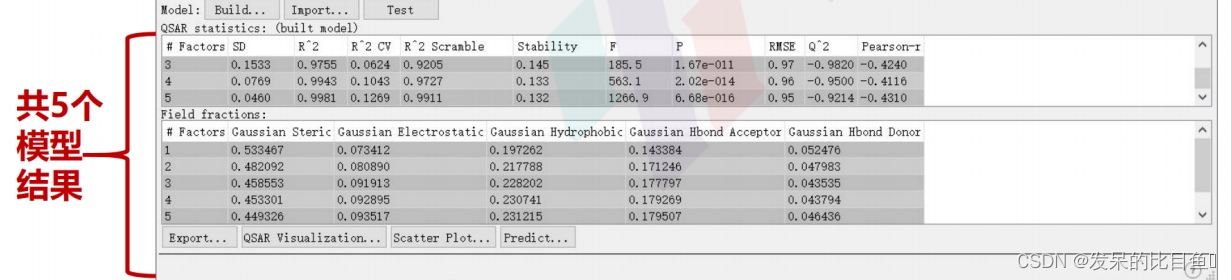
√
q
2
q^2
q2(Cross-validated r2cv):交叉验证系数,用于评价模型的主要参数
√
q
2
>
0.50
q^2>0.50
q2>0.50:有统计显著性的预测模型
√
q
2
>
0.4
q^2>0.4
q2>0.4:可以考虑使用该模型
√
q
2
<
0
q^2<0
q2<0:模型预测能力低于以均值预测能力
此处,我们选择第4个模型进行后续分析
3DQSAR的统计参数
ColumnDescription#Factors偏最小二乘回归模型中的因子数。SD回归的标准差。这是拟合的活性值的RMS误差,分布在n-m-1个自由度(n个配体,m个PLS因子)上。R^2回归的标准差。这是拟合的活性值的RMS误差,分布在n-m-1个自由度(n个配体,m个PLS因子)上。R^2 CV回归的R值(决定系数)。例如,0.80的值意味着模型占观测活动数据方差的80%。R始终在0和1之间。R^2 Scramble使用扰乱活动构建的一系列模型的R平均值。衡量分子场能够拟合随机数据的程度。低值意味着模型不能拟合随机数据,但高值只意味着变量集相当完整,可以拟合任何东西。Stability模型预测对训练集组成变化的稳定性。最大值为1。高值表示模型对训练集中的遗漏不敏感。稳定性值低于R-值表示过度拟合。F模型方差与观测到的活动方差之比。模型方差分布在m个自由度上,活性方差分布在n-m-1个自由度(n个配体,mPLS因子)上。F的大值表明回归具有更大的统计学意义。P将F作为卡方分布的比率处理时的显著性水平。值越小表示置信度越高。P值为0.05意味着F在95%的水平上是显著的。RMSE测试集预测中的均方根误差。Q^2预测活动的Q值。直接类似于R平方,但基于测试集预测。如果误差的方差大于观察到的方差,Q可以取负值Pearson-r测试集预测活动与观察活动之间相关性的Pearson-r值。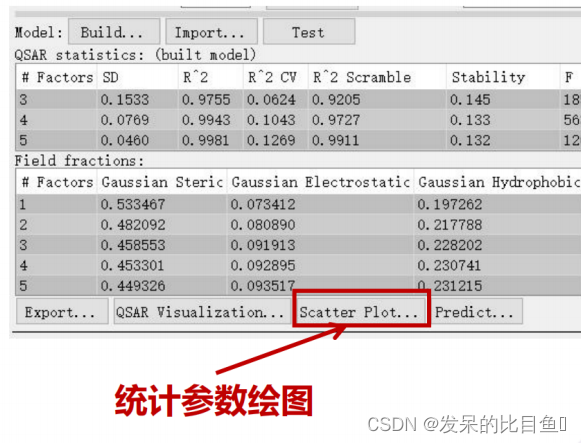
预测值和实际值的相关性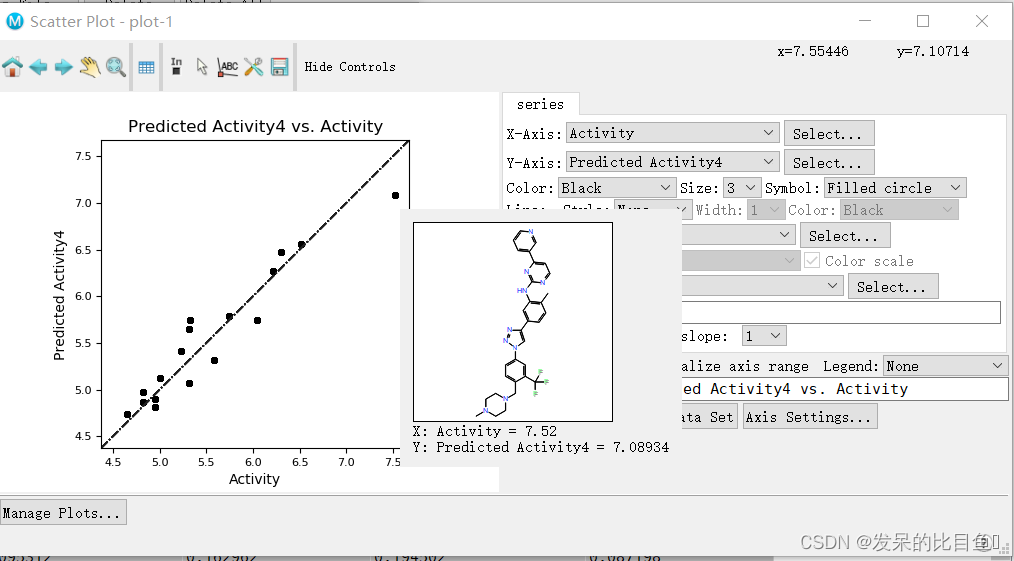
训练集:预测值和实际值的相关性(调整参数根据活性大小区分颜色)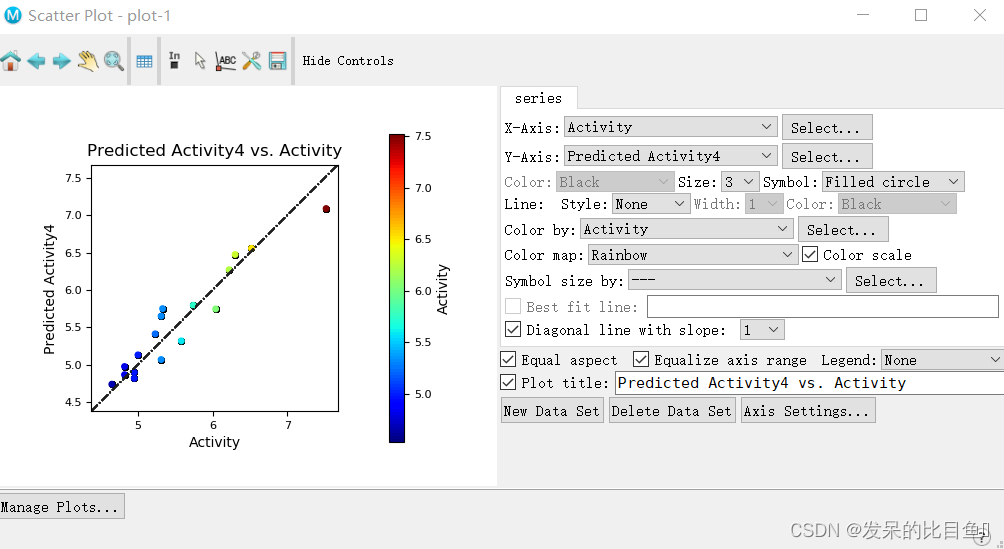
测试集:预测值和实际值的相关性(调整参数根据活性大小区分颜色)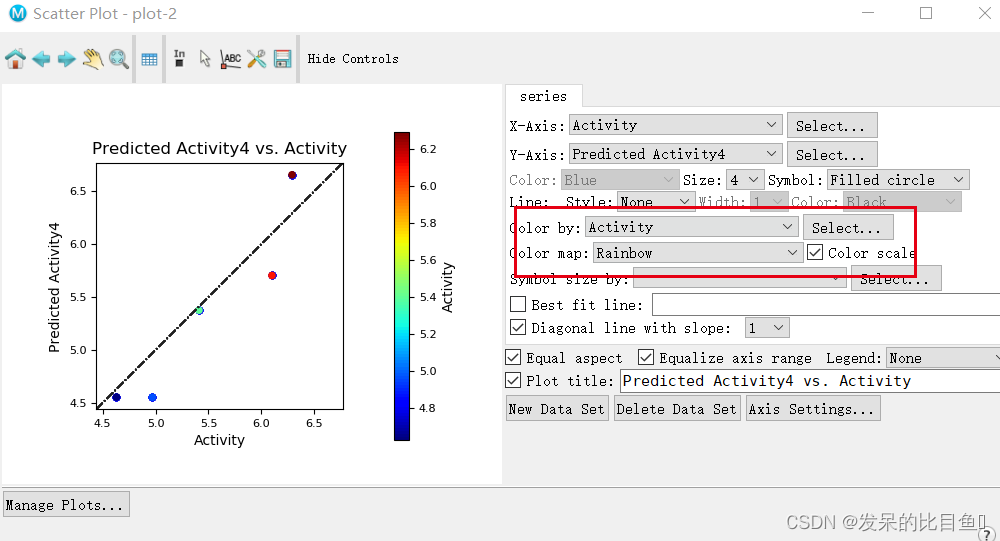


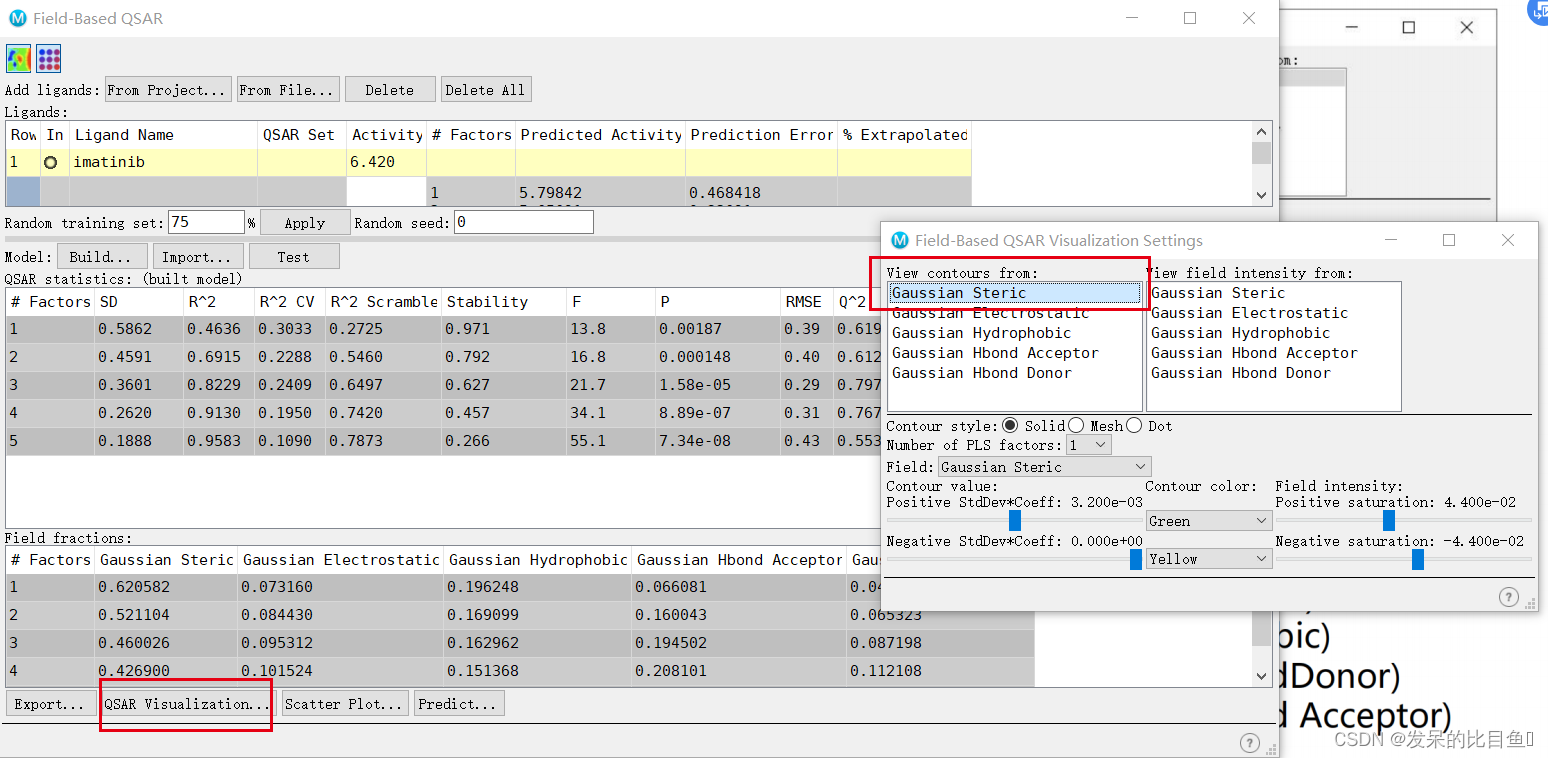
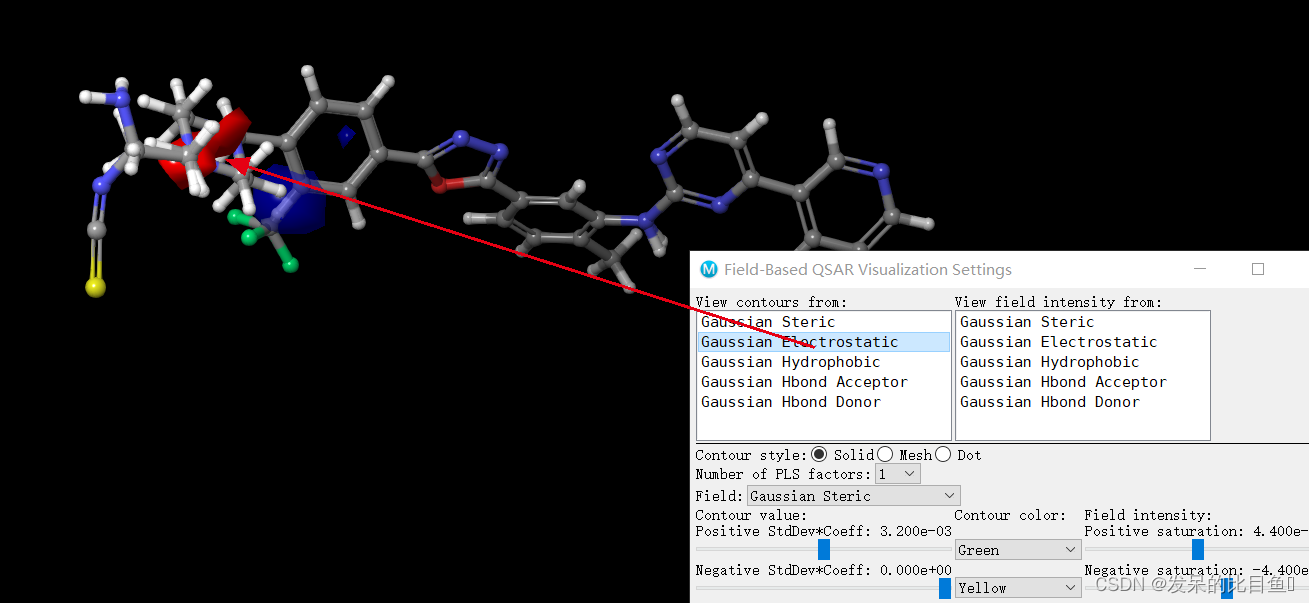
3D-QSAR模型提供了五个场的场信息,分别为:
- 立体场(Steric)
- 静电场(Electrostatic)
- 疏水场(Hydrophobic)
- 氢键供体场(HbondDonor)
- 氢键受体场(Hbond Acceptor)
等势能图解读


- 黄绿色块表示立体场等势能图: - 绿色表示该处引入大基团即增大体积会增加活性- 黄色表示该处引入小基团即减小体积有利于提高活性
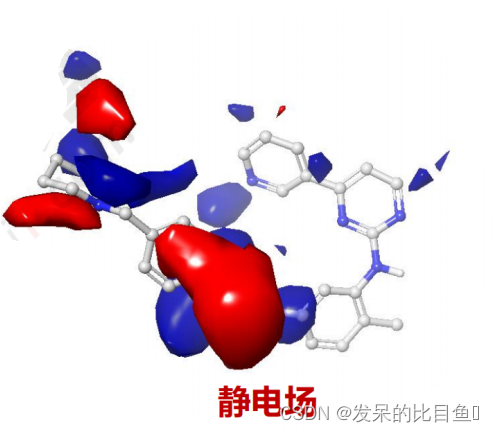
- 红蓝色块表示静电场等势能图: - 蓝色表示增加正电荷有利于增强活性- 红色表示增加负电荷有利于增强活性
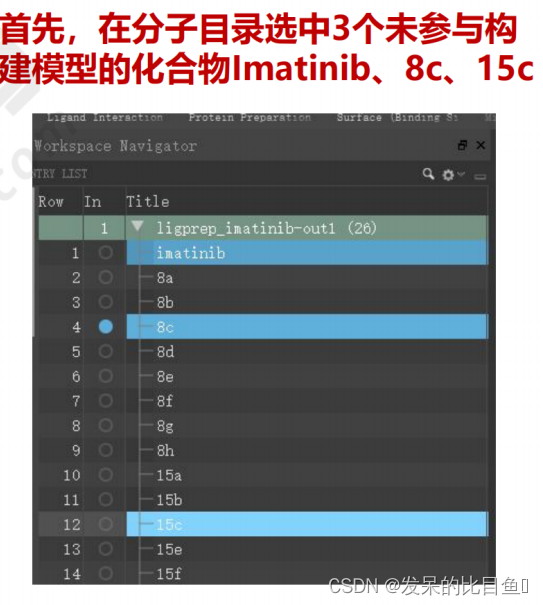
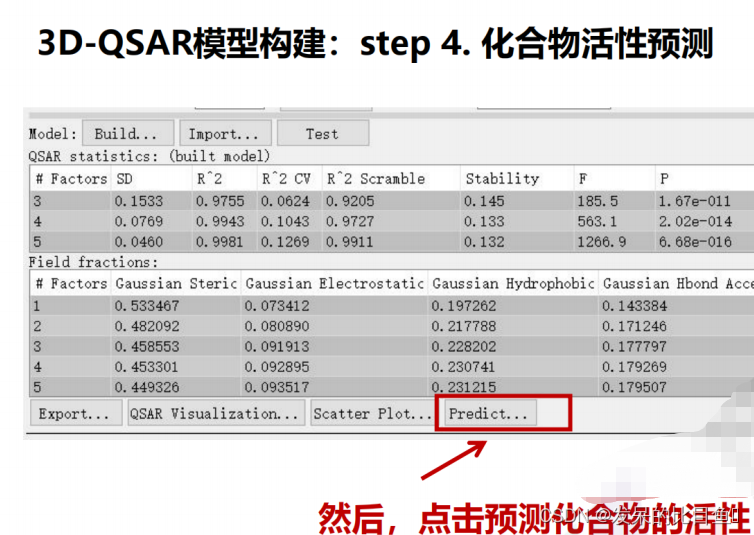
模型调整策略
- 能量优化:更改优化的参数比如立场、梯度、优化次数,添加电荷的类型等
- 能量优化是为了尽可能接近化合物的活性构象,因此可以将化合物与靶点蛋白对接,取其对接构象
- 分子叠合:模型的优劣很大程度取决于叠合的好坏,因此需要尝试不同的叠合方式,必要时可删除不合适的分子
版权归原作者 发呆的比目鱼 所有, 如有侵权,请联系我们删除。