
在这篇文章中,我们将比较LASSO、PLS、Random Forest等多变量模型与单变量模型的预测能力,如著名的差异基因表达工具DESeq2以及传统的Mann-Whitney U检验和Spearman相关。使用骨骼肌RNAseq基因表达数据集,我们将展示使用多变量模型构建的预测得分,以优于单变量特征选择模型。
骨骼肌RNAseq基因表达数据
在这里,我们将量化几种特征选择方法的预测能力:a)单变量(逐个)特征选择,b)多变量(一起)特征选择。出于演示目的,我们将使用来自GTEX人体组织基因表达联盟的骨骼肌RNAseq基因表达数据集(为简单起见,随机抽取1000个基因)。
版本6的GTEX骨骼肌数据集包括157个样本。我们装载基因表达矩阵X,去除低表达基因。为了与组学整合的后选择特征相一致,我们将使用性别作为我们将要预测的响应变量Y。让我们通过降维(如PCA)来快速可视化这些样本。
library("mixOmics")
X<-read.table("GTEX_SkeletalMuscles_157Samples_1000Genes.txt",
header=TRUE,row.names=1,check.names=FALSE,sep="\t")
X<-X[,colMeans(X)>=1]
Y<-read.table("GTEX_SkeletalMuscles_157Samples_Gender.txt",
header=TRUE,sep="\t")$GENDER
names(Y)<-rownames(X)
pca.gtex<-pca(X, ncomp=10)
plotIndiv(pca.gtex, group=Y, ind.names=FALSE, legend=TRUE)
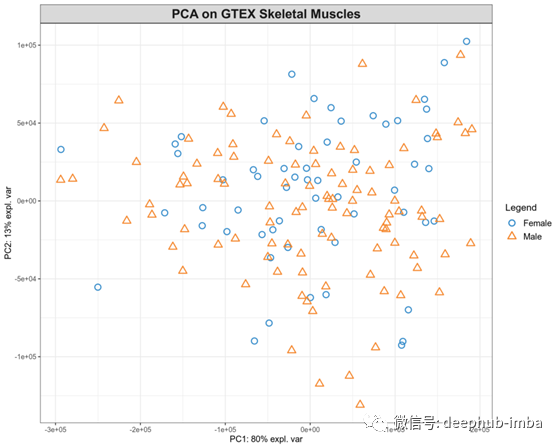
正如我们所看到的,PCA图并没有根据骨骼肌基因表达数据显示出男性和女性之间的明显区别。我们只能假设基因性染色体(X, Y)可以提供一个完美的男性和女性之间的分离,但是我们没有观察到这种情况,因为:1)大多数基因来自常染色体,2)随机抽样 大约有1000个基因,因此我们可能没有从X和Y染色体上取样许多基因。在下一节中,我们将把数据集分割成训练和测试子集,然后在训练集上实现单变量和多变量特征选择(训练)模型,并使用平衡假阳性(FPR)和真阳性(TPR)率的roc曲线技术对测试集上的模型进行评估。
性别预测:LASSO与单变量方法
为了评估单变量和多变量模型的预测能力,我们需要对独立的数据集进行训练和评估。为此,让我们将数据集划分为训练(80%的样本)和测试(20%的样本)子集。为了确保我们的评估不基于数据的特定“幸运”拆分,我们将多次对训练和测试子集执行随机拆分,多次计算每次ROC曲线并平均多个ROC曲线。这样,我们将一举两得:为每个模型的ROC曲线建立置信区间,并使ROC曲线平滑且美观,否则,除非您在测试中有大量样本,否则它们将会出现各种问题。
在本篇文章中,我将添加另一种非ivarite特征选择方法,即Mann-Whitney U检验,该检验应在很大程度上与Spearman相关性相比较,因为两者都是非参数和基于秩的单变量方法,因此不假定数据的特定分布。为了将通过单变量方法单独选择的基因组合到预测得分中,我们将使用它们的表达与性别之间的个体关联性的p值对它们进行排名,并通过Bonferroni程序校正多次测试。经过多次测试调整后,这通常会在训练数据集上产生一些差异表达的基因。此外,可以通过计算其在训练数据集上的作用/权重的乘积(Spearman rho和男性与女性之间基因表达的倍数变化的对数),将差异显着表达(男性与女性之间)的基因折叠成预测得分。以及来自测试数据集的样本的基因表达值。让我们对其进行编码,并比较模型之间的ROC曲线。
roc_obj_univar_FPR_list<-list();roc_obj_univar_TPR_list<-list()
roc_obj_wilcox_univar_FPR_list<-list();roc_obj_wilcox_univar_TPR_list<-list()
roc_obj_multivar_FPR_list<-list();roc_obj_multivar_TPR_list<-list()
roc_obj_univar_AUC<-vector();roc_obj_wilcox_univar_AUC<-vector()
roc_obj_multivar_AUC<-vector()
for(jin1:100)
{
#RANDOMLY SPLIT DATA SET INTO TRAIN AND TEST
test_samples<-rownames(X)[sample(1:length(rownames(X)),
round(length(rownames(X))*0.2))]
train_samples<-rownames(X)[!rownames(X)%in%test_samples]
X.train<-X[match(train_samples,rownames(X)),]
X.test<-X[match(test_samples,rownames(X)),]
Y.train<-Y[match(train_samples,names(Y))];Y.test<-Y[match(test_samples,names(Y))]
#TRAIN AND EVALUATE MULTIVARIATE LASSO MODEL
lasso_fit<-cv.glmnet(as.matrix(X.train), Y.train,family="binomial",alpha=1)
coef<-predict(lasso_fit, newx=as.matrix(X.test), s="lambda.min")
roc_obj_multivar<-rocit(as.numeric(coef),Y.test)
#TRAIN AND EVALUATE UNIVARIATE SPEARMAN CORRELATION MODEL
rho<-vector();p<-vector()
for(iin1:dim(X.train)[2])
{
corr_output<-cor.test(X.train[,i],as.numeric(Y.train),method="spearman")
rho<-append(rho,as.numeric(corr_output$estimate))
p<-append(p,as.numeric(corr_output$p.value))
}
output_univar<-data.frame(GENE=colnames(X.train),SPEARMAN_RHO=rho,PVALUE=p)
output_univar$FDR<-p.adjust(output_univar$PVALUE,method="BH")
output_univar<-output_univar[order(output_univar$FDR,output_univar$PVALUE,
-output_univar$SPEARMAN_RHO),]
output_univar<-output_univar[output_univar$FDR<0.05,]
my_X<-subset(X.test,select=as.character(output_univar$GENE))
score<-list()
for(iin1:dim(output_univar)[1])
{
score[[i]]<-output_univar$SPEARMAN_RHO[i]*my_X[,i]
}
roc_obj_univar<-rocit(colSums(matrix(unlist(score), ncol=length(Y.test),
byrow=TRUE)),Y.test)
#TRAIN AND EVALUATE MANN-WHITNEY U TEST UNIVARIATE MODEL
wilcox_stat<-vector();p<-vector();fc<-vector()
for(iin1:dim(X.train)[2])
{
wilcox_output<-wilcox.test(X.train[,i][Y.train=="Male"],
X.train[,i][Y.train=="Female"])
wilcox_stat<-append(wilcox_stat,as.numeric(wilcox_output$statistic))
fc<-append(fc,mean(X.train[,i][Y.train=="Male"])/mean(
X.train[,i][Y.train=="Female"]))
p<-append(p,as.numeric(wilcox_output$p.value))
}
output_wilcox_univar<-data.frame(GENE=colnames(X.train),MWU_STAT=wilcox_stat,
FC=fc,PVALUE=p)
output_wilcox_univar$LOGFC<-log(output_wilcox_univar$FC)
output_wilcox_univar$FDR<-p.adjust(output_wilcox_univar$PVALUE,method="BH")
output_wilcox_univar<-output_wilcox_univar[order(output_wilcox_univar$FDR,
output_wilcox_univar$PVALUE),]
output_wilcox_univar<-output_wilcox_univar[output_wilcox_univar$FDR<0.05,]
my_X<-subset(X.test,select=as.character(output_wilcox_univar$GENE))
score<-list()
for(iin1:dim(output_wilcox_univar)[1])
{
score[[i]]<-output_wilcox_univar$LOGFC[i]*my_X[,i]
}
roc_obj_wilcox_univar<-rocit(colSums(matrix(unlist(score),ncol=length(Y.test),
byrow=TRUE)),Y.test)
#POPULATE FPR AND TPR VECTORS / MATRICES
roc_obj_univar_FPR_list[[j]]<-roc_obj_univar$FPR
roc_obj_univar_TPR_list[[j]]<-roc_obj_univar$TPR
roc_obj_wilcox_univar_FPR_list[[j]]<-roc_obj_wilcox_univar$FPR
roc_obj_wilcox_univar_TPR_list[[j]]<-roc_obj_wilcox_univar$TPR
roc_obj_multivar_FPR_list[[j]]<-roc_obj_multivar$FPR
roc_obj_multivar_TPR_list[[j]]<-roc_obj_multivar$TPR
roc_obj_univar_AUC<-append(roc_obj_univar_AUC,roc_obj_univar$AUC)
roc_obj_wilcox_univar_AUC<-append(roc_obj_wilcox_univar_AUC,
roc_obj_wilcox_univar$AUC)
roc_obj_multivar_AUC<-append(roc_obj_multivar_AUC,roc_obj_multivar$AUC)
print(paste0("FINISHED ",j,"ITERATIONS"))
}
#AVERAGE MULTIPLE ROC-CURVES OR EACH MODEL AND PLOT MEAN ROC-CURVES
roc_obj_univar_FPR_mean<-colMeans(matrix(unlist(roc_obj_univar_FPR_list),
ncol=length(Y.test)+1,byrow=TRUE))
roc_obj_univar_TPR_mean<-colMeans(matrix(unlist(roc_obj_univar_TPR_list),
ncol=length(Y.test)+1,byrow=TRUE))
roc_obj_wilcox_univar_FPR_mean<-colMeans(matrix(
unlist(roc_obj_wilcox_univar_FPR_list), ncol=length(Y.test)+1,byrow=TRUE))
roc_obj_wilcox_univar_TPR_mean<-colMeans(matrix(
unlist(roc_obj_wilcox_univar_TPR_list), ncol=length(Y.test)+1,byrow=TRUE))
roc_obj_multivar_FPR_mean<-colMeans(matrix(unlist(roc_obj_multivar_FPR_list),
ncol=length(Y.test)+1,byrow=TRUE))
roc_obj_multivar_TPR_mean<-colMeans(matrix(unlist(roc_obj_multivar_TPR_list),
ncol=length(Y.test)+1,byrow=TRUE))
plot(roc_obj_univar_FPR_mean,roc_obj_univar_TPR_mean,col="green",
ylab="SENSITIVITY (TPR)",xlab="1 - SPECIFISITY (FPR)",cex=0.4,type="o")
lines(roc_obj_multivar_FPR_mean,roc_obj_multivar_TPR_mean,cex=0.4,col="blue",
type="o",lwd=2)
lines(roc_obj_wilcox_univar_FPR_mean,roc_obj_wilcox_univar_TPR_mean,cex=0.4,
col="red",type="o",lwd=2)
lines(c(0,1),c(0,1),col="black")
legend("bottomright", inset=.02,
c("Multivarite: LASSO","Univarite: SPEARMAN",
"Univarite: MANN-WHITNEY U TEST"), fill=c("blue","green","red"))

在这里我们可以得出结论,LASSO比这两种单变量特征选择方法具有更大的预测能力。更好看的差异ROC曲线下的面积(AUC ROC)之间的三种方法,以及能够执行统计测试来解决如何重要ROC曲线之间的差异,让我们做一个箱线图的AUC ROC套索,单变量斯皮尔曼相关和Mann-Whitney U测试。
boxplot(roc_obj_multivar_AUC,roc_obj_univar_AUC,roc_obj_wilcox_univar_AUC,
names=c("LASSO","SPEARMAN","MANN-WHITNEY U TEST"),col=c("blue","green","red"))
mwu<-wilcox.test(roc_obj_wilcox_univar_AUC,roc_obj_multivar_AUC)
mtext(paste0("LASSO vs. Mann-Whitney U test: P-value = ",mwu$p.value))
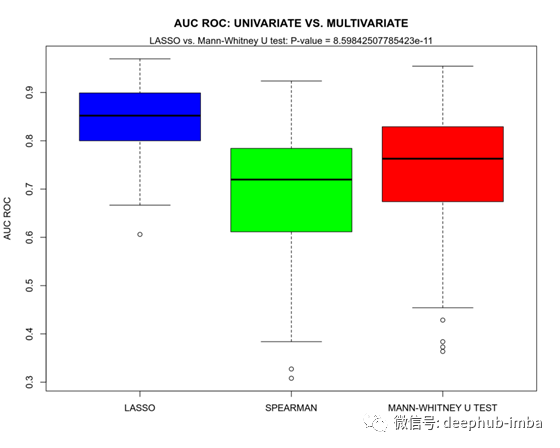
我们可以看到,Spearman correlation和Mann-Whitney U test单变量特征选择模型具有相当的AUC ROC指标(尽管Mann-Whitney U test较好),且两者的AUC ROC即预测能力均明显低于multivarite LASSO。
然而,这种比较可能存在偏差。谈到单变量模型(Spearman和Mann-Whitney U检验),我们提到只有少数基因在多次检验的Bonferroni校正后是显著的。所以我们在构建单变量模型的预测得分时只使用了少量的基因,而LASSO选择了更多的基因30个,请参阅github上的完整代码。如果LASSO更好的预测能力仅仅是因为它的预测分数使用了更多的特征呢?为了验证这一假设,在下一节中,我们将暂时忽略Bonferroni校正,并使用Spearman相关性和Mann-Whitney U检验,单独使用p值排序来确定30个最具预测性的基因。换句话说,我们将使用与多品种相同数量的基因来构建其预测得分。通过选择的基因数量来模拟LASSO的相应的单变量模型称为SPEAR30 (Spearman correlation with ~30 differential expressed genes)和MWU30 (Mann-Whitney U test with ~30 differential expressed genes)。
性别预测:DESeq2与多元方法
在本节中,除了将LASSO与SPEAR30(具有约30个差异表达基因的Spearman相关性)模型和MWU30(具有约30个差异表达基因的Mann-Whitney U检验)模型进行比较之外,我们还将添加其他一些流行的单变量和多变量模型。
首先,当进行差异基因表达分析时,DESeq2是业界的一个黄金标准。该工具享有很高的声誉,在RNAseq社区中非常受欢迎。这是一个单变量工具,即假设基因表达计数为负二项分布,它会执行逐个基因的测试。此外,它采用了方差稳定程序,其中高表达的基因有助于低表达的基因得到正确测试。比较DESeq2预测能力与Mann-Whitney U检验和Spearman相关性,这些检验本质上利用相同的单变量思想,但是与假定基因表达为负生物学分布的DESeq2相比,都执行非参数类型的检验,这将是很有意思的。执行参数测试。并行地,我们将计算DESEQ2_30模型,该模型使用与LASSO选择的相同数目的基因(按与性别相关的p值排序)建立预测得分。类似于SPEAR30和MWU30模型。
其次,我们将添加另外两个多元特征选择模型,以与LASSO和单变量模型进行比较。这两个是偏最小二乘判别分析(PLS-DA)和随机森林,它们都是通用的多元模型。其中一个(PLS-DA)和LASSO是线性的,另一个(Random Forest)是非线性的。在这里,我们不仅旨在比较单变量或多变量特征选择模型,而且还想了解与线性LASSO和PLS-DA相比,非线性随机森林能否改善预测。
如上一节所述,我们将通过将数据集分为多次训练和测试来围绕ROC曲线建立置信区间。它涉及很多编码,我在这里不介绍代码,但是欢迎您在我的github上检查它们。在下面,我仅介绍经过多次训练测试后的每个模型的平均ROC曲线,以及AUC的箱线图。
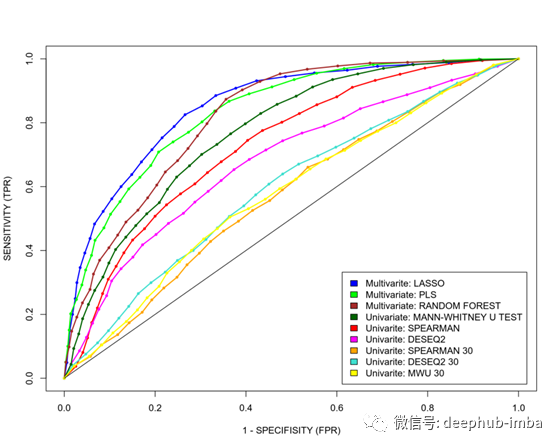
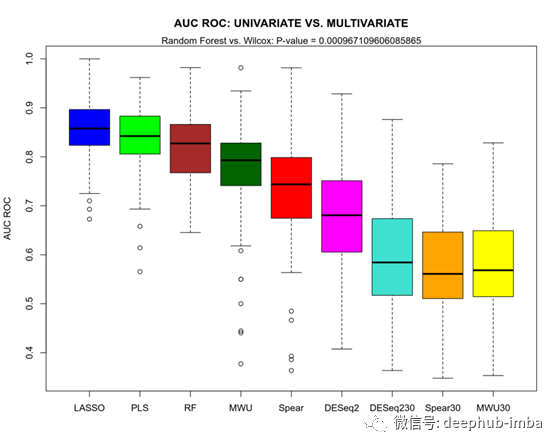
我们在这里观察到一些有趣的事情。首先,与所有多变量模型相比,所有单变量模型的预测能力似乎都更差。即使是最差的多元模型,在这里是随机森林(RF),其AUC ROC也明显高于最好的单变量模型,在这里似乎是Mann-Whitney U检验(MWU)。
其次,具有与LASSO选择的基因数量相同的所有单变量模型(DESeq230,SPEAR30和MWU30)无法与所有其他单变量或多变量模型竞争,这暗示单变量模型的预测能力较差的原因不是由于数目不同特征/基因的选择,但由于构建预测得分的基因的等级和权重不同而异。
第三,与线性多变量LASSO和PLS-DA模型相比,非线性多变量随机森林对RNAseq基因表达的预测效果似乎没有改善。然而,根据我的经验,许多生命科学问题通常都是这样,在使用非线性分类器之前,通常值得检查一下简单的线性模型。
第四,也是最有趣的是,DESeq2单变量参数预测得分似乎不仅比多变量模型(LASSO, PLS-DA, Random Forest)表现更差,而且比单变量非参数模型,如Spearman相关和Mann-Whitney U检验也差。考虑到上述非参数测试的简单性和DESeq2的卓越声誉,这是相当出乎意料的。然而,事实证明,至少对于这个特定的数据集,简单的Spearman和Mann-Whitney非参数测试在预测能力方面优于DESeq2。
LASSO,Ridge和Elastic Net
作为奖励,在本节中,我们将比较LASSO(L1规范),Ridge(L2规范)和Elastic Net(L1和L2规范的组合)的预测能力的预测得分。通常,惩罚线性模型族的这三个成员之间的区别并不明显。在不赘述的情况下(有很多文献解释了这种差异),我们仅强调LASSO是最保守的方法,因此由于生命科学数据的高噪声水平,在生命科学中通常首选LASSO。Elastic Net在Ridge的凸优化优势和LASSO的严格性之间提供了很好的平衡,并且在癌症研究中非常受欢迎。接下来,我们再次对RNAseq基因表达数据集进行100次训练测试后,得出LASSO,Ridge和Elastic Net的平均ROC曲线,以及AUC ROC指标的箱线图。
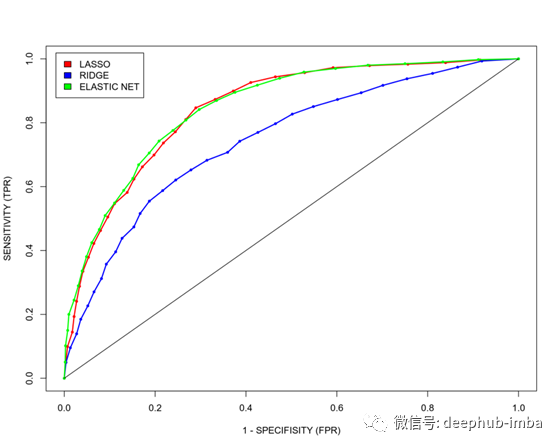

我们可以看到LASSO和Elastic Net给出了几乎相同的ROC曲线,并且胜过了已知的最宽松的Ridge模型,这显然不利于模型概括,因此对于预测目的无益,因此Ridge可能不是第一个生命科学数据的最佳选择。
总结
在本文中,我们了解到,与单变量模型相比,多元统计模型似乎具有更好的预测能力。至少对于本文研究的GTEX骨骼肌RNAseq基因表达数据而言,单变量差异基因表达工具DESeq2不仅比诸如LASSO,PLS-DA和Random Forest等多变量模型,而且与简单非 参数单变量模型,例如Spearman相关性和Mann-Whitney U检验。
本文代码:https://github.com/NikolayOskolkov/UnivariteVsMultivariteModels
作者:Nikolay Oskolkov
原文地址:https://towardsdatascience.com/univariate-vs-multivariate-prediction-c1a6fb3e009
deephun翻译组