
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档
文章目录
一、STAR比对
首先在NCBI上下拟南芥基因组序列(fasta格式)文件和基因组注释文件(GTF格式)。
开启共享文件夹功能,在windows系统中将下载的文件放入共享文件夹中,共享文件位置在/mnt/hgfs/fold name,fold name即在windows系统中共享文件夹的名称。
1.建立基因组索引
使用STAR进行基因组索引拟南芥基因组序列(fasta/fna格式,fna文件可直接改名fa)文件和基因组注释文件(GTF格式)。
确保提前进入rna环境。该程序会比较吃内存,保证虚拟机内存至少为8G.
conda activate rna
STAR --runMode genomeGenerate --genomeDir /home/lumino/TEST1/STARoutput2 --genomeFastaFiles /home/lumino/TEST1/GCA_000001735.2_TAIR10.1_genomic.fa --sjdbGTFfile /home/lumino/TEST1/genomic.gtf --sjdbOverhang 99 --genomeSAindexNbases 12
STAR:这是运行 STAR 工具的命令。--runMode genomeGenerate:这是指定 STAR 运行的模式,即生成基因组索引。--/home/lumino/TEST1/STARoutput2:这是指定生成的基因组索引文件的输出目录路径。您需要将/home/lumino/TEST1/STARoutput2替换为您希望保存索引文件的实际路径。--genomeFastaFiles /home/lumino/TEST1/GCA_000001735.2_TAIR10.1_genomic.fa:这是指定包含基因组序列的 FASTA 文件的路径。您需要将/home/lumino/TEST1/GCA_000001735.2_TAIR10.1_genomic.fa替换为您实际的基因组序列文件路径。--sjdbGTFfile /path/to/annotation.gtf:这是指定包含注释信息的 GTF 文件的路径。您需要将/home/lumino/TEST1/genomic.gtf替换为您实际的注释文件路径。--sjdbOverhang 99:这是指定用于比对的 reads 的长度减去 1。在这个例子中,100表示 reads 的长度减去 1 为 99。这个参数的设置会影响 STAR 的比对效果。- --genomeSAindexNbases 12 推荐的参数,默认为14,但可能对这个基因组大小来说太大。
- 完成后在文件夹内生以下文件。
2.作图
使用STAR对SRR3418005-filtered.fastq文件进行比对和基因组定位。
确保内存大于12G(很重要),新建一个STAR_result的文件夹。
STAR --genomeDir /home/lumino/TEST1/STARoutput2 --readFilesIn /home/lumino/TEST1/SRR3418005-filtered.fastq --runThreadN 4 --outSAMstrandField intronMotif --sjdbGTFfile /home/lumino/TEST1/genomic.gtf --outFileNamePrefix /home/lumino/TEST1/STARoutput2/STAR_result/SRR3418005-filtered-STAR
--genomeDir /home/lumino/TEST1/STARoutput2: 指定STAR索引文件的目录路径。--readFilesIn /home/lumino/TEST1/SRR3418005-filtered.fastq: 指定要比对的fastq文件路径。--runThreadN 4: 指定使用的线程数为4,用于加速比对过程。--outSAMstrandField intronMotif: 指定输出的SAM文件中包含intron motif信息。--sjdbGTFfile /home/lumino/TEST1/genomic.gtf: 指定用于注释的GTF文件路径。--outFileNamePrefix /home/lumino/TEST1/STARoutput2/STAR_result/SRR3418005-filtered-STAR: 指定输出文件的前缀和路径。
- 产以下文件。

3.SAM文件处理
以BAM格式存储比对节省空间,并且许多下游工具使用BAM格式,而不是SAM。
使用samtools将SAM文件转换为BAM。去报提前下载samtools。
cd /home/lumino/TEST1/STARoutput3/STAR_result
#进入之前STAR 产生SAM文件夹
samtools view -b SRR3418005-filtered-STARAligned.out.sam | samtools sort -o SRR3418005-filtered-STAR.sorted.bam
#对SRR3418005建立索引,输出bai文件
samtools index SRR3418005-filtered-STAR.sorted.bam SRR3418005.bai
samtools view -b SRR3418005-filtered-STARAligned.out.sam:这部分命令将输入的SAM文件SRR3418005-filtered-STARAligned.out.sam转换为BAM格式。samtools view命令用于查看和转换SAM/BAM格式文件,-b选项表示输出为BAM格式。|(Enter键上方,shift和\键一起按):这个符号表示管道,将第一个命令的输出传递给下一个命令作为输入。samtools sort -o SRR3418005-filtered-STAR.sorted.bam:这部分命令将通过管道接收到的BAM文件进行排序,并将排序后的结果保存为SRR3418005-filtered-STAR.sorted.bam文件。samtools sort命令用于对BAM文件进行排序,-o选项表示指定输出文件名。
4.比对结果可视化
使用IGV展示比对结果,导入基因组
GCA_000001735.2_TAIR10.1_genomic.fa和SRR3418005-filtered-STAR.sorted.bam。
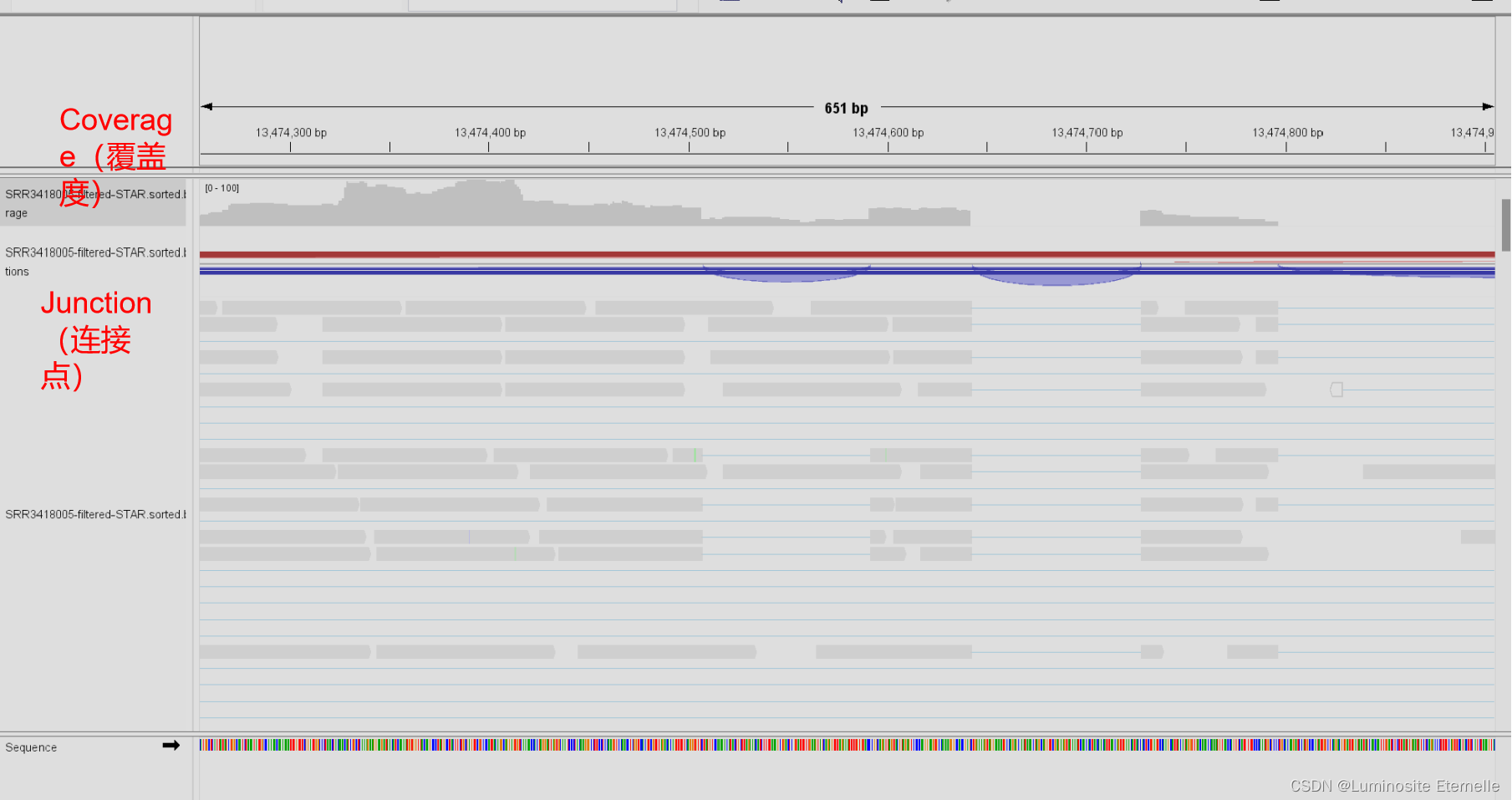
- Coverage(覆盖度):Coverage指的是在RNA测序数据中每个碱基被测序的次数或覆盖的深度。在转录组测序数据中,coverage可以用来衡量每个基因或转录本的表达水平。高coverage表示该基因或转录本在样本中具有较高的表达水平,而低coverage则表示较低的表达水平。通过分析coverage可以帮助研究人员了解基因的表达情况、差异表达和基因调控等信息。
- Junction(连接点):Junction指的是RNA测序数据中的剪接位点,即不同外显子之间的连接点。在转录组测序数据中,剪接是指在RNA后转录过程中,不同外显子之间的连接方式。Junction信息可以帮助研究人员识别和分析基因的剪接事件,包括已知的和新发现的剪接形式。通过分析junction可以揭示基因的剪接异构体、剪接调控机制等重要信息。
5.比对质量评估
qualimap bamqc -bam SRR3418005-filtered-STAR.sorted.bam -outdir ./ -gff ./genomic.gtf -nt 4
-bam SRR3418005-filtered-STAR.sorted.bam: 指定输入的BAM文件,这里是一个名为SRR3418005-filtered-STAR.sorted.bam的文件,它可能已经过滤(filtered)和排序(sorted)。-outdir ./: 指定输出目录,.表示当前目录。qualimap将在该目录下生成各种统计文件和图表。-gff ./genomic.gtf: 使用一个GFF(General Feature Format)或GTF(Gene Transfer Format)文件来注释基因组特征。这里使用的是./genomic.gtf文件,它包含了基因、转录本等基因组注释信息。这些信息对于评估测序数据在基因区域的覆盖情况和分布非常重要。-nt 4: 指定使用的线程数(number of threads),这里是4个线程。多线程可以加速计算过程,特别是在处理大型数据集时。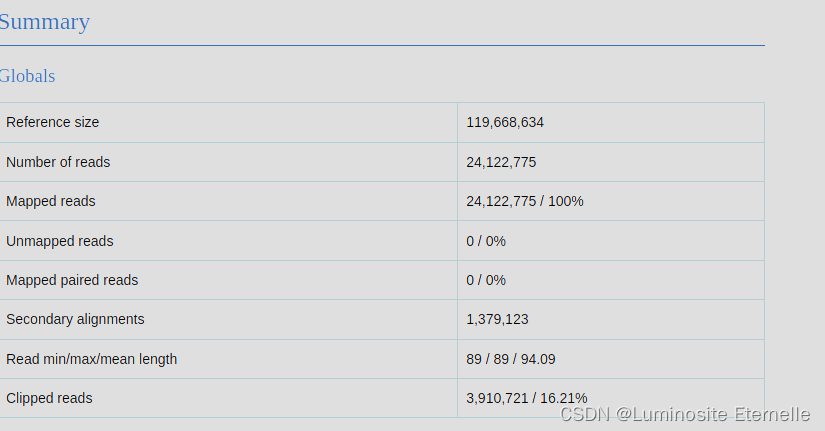
- 映射率:所有读取(reads)都成功地映射到了参考基因组上,映射率为100%。这是一个非常理想的结果,意味着所有的测序数据都能与参考序列匹配,从而能够用于后续的分析。
- 未映射读取:没有未映射的读取,这进一步验证了数据的高质量和完整性。
- 配对读取映射:由于没有配对读取(Mapped paired reads 为 0),这可能表明这是单端测序数据或数据处理过程中没有保留配对信息。对于单端测序,这不影响数据的可用性,但对于配对端测序,则可能表明数据预处理存在问题。
- 次级对齐:有1,379,123个读取存在次级对齐。次级对齐通常发生在读取来自基因组中的重复区域或高度相似区域时。这些读取可能不是唯一地映射到参考序列上,但这并不一定意味着它们的质量差,只是表明这些区域在基因组中是复杂的。
- 读取长度:读取的最小长度是89bp,最大长度也是89bp,说明所有读取的长度都是一致的,这通常是一个好的迹象。平均读取长度是94.09bp,稍微超过了最小长度,可能是因为有些读取在质量控制或修剪过程中被轻微缩短了。
- 修剪的读取:有3,910,721个读取被修剪了,占总读取数的16.21%。这可能是由于测序质量不佳、含有接头序列或其他原因导致的。修剪可能会影响后续分析的准确性,但具体影响取决于修剪的程度和原因。
- 综合以上分析,我们可以认为这些测序数据在映射率方面表现非常好,但存在一定数量的次级对齐和修剪的读取。为了更全面地评估数据质量,您可能需要进一步查看修剪的读取的比例和分布,以及次级对齐的具体原因。此外,进行碱基质量分布、GC含量等其他质量指标的分析也是很有必要的。这些都可以通过专业的质量控制工具(如FastQC)来实现。

基于您提供的全局统计数据,我们可以对测序数据在特定区域内的质量进行以下评估:
区域大小和覆盖:
- 特定区域的大小为76,556,503,占参考序列的63.97%,这表示覆盖了大部分参考基因组。
- 映射的读取数为24,047,087,占所有读取的99.69%,这是一个非常高的映射率,表明大多数测序数据都成功匹配到了参考序列。
重复读取:
- 估计的重复读取数为17,478,788,占所有映射读取的72.69%。这意味着大约三分之二的读取是重复的,可能是由于PCR扩增或其他测序过程中的问题导致的。
- 重复率为45.08%,这是一个相对较高的重复率,可能会影响后续的变异检测、基因表达分析等。在后续分析中,可能需要对数据进行去重复处理。
碱基组成:
- A、C、T、G四种碱基的比例分别为27.71%、23.69%、27.06%和21.53%,整体分布较为均衡,没有明显的碱基偏倚。
- GC含量为45.23%,这也是一个正常的范围,没有显示出异常的GC含量。
- N的数量非常少(42,487),仅占所有碱基的极小比例,这通常意味着测序质量较好,没有太多的不确定碱基。
结论:
- 整体来看,测序数据在特定区域内的映射率很高,但重复率也相对较高。碱基组成分布均衡,没有明显的偏倚。
- 在后续分析中,建议对重复读取进行去重处理,以提高分析的准确性。同时,考虑到高重复率可能掩盖了真实的生物学信号,需要谨慎解释这些结果,并在可能的情况下使用其他技术或方法验证数据。
覆盖率评估:
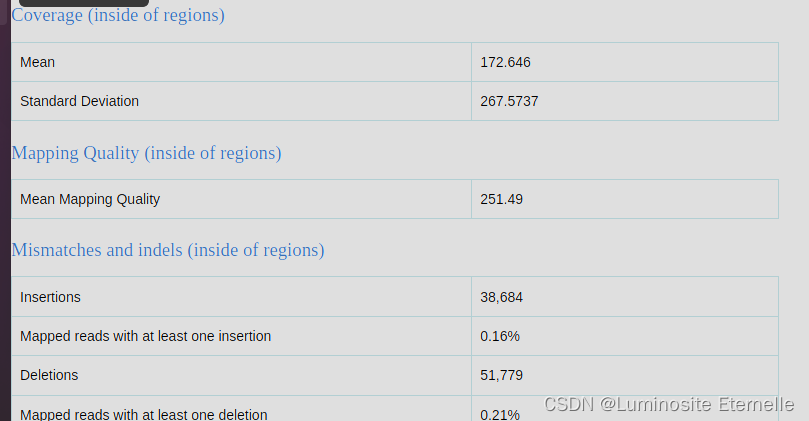
- 平均覆盖率为172.646倍,这表示每个碱基平均被测序了超过172次。这个深度通常是非常高的,对于大多数类型的分析,包括变异检测、基因表达定量等,都足够了。
- 覆盖率的标准差为267.5737,表明覆盖深度在不同区域之间存在一定的波动。这种波动可能是由于测序技术本身的局限性、基因组不同区域的特性,或实验条件的不同所致。尽管标准差相对较高,但平均覆盖率仍然很高,因此这种波动可能不会对大多数分析产生太大影响。
映射质量评估:
- 平均映射质量为251.49,这是一个非常高的值,意味着大多数读取都能非常准确地映射到参考序列上。这通常意味着测序数据的质量很好,可以信赖。
插入和删除事件:
- 插入事件总数为38,684次,占映射读取的0.16%。这个比例相对较低,表明测序过程中插入错误的发生频率并不高。
- 删除事件总数为51,779次,占映射读取的0.21%。与插入事件相似,删除事件的比例也较低,说明测序数据在删除错误方面也相对可靠。
同源聚合物插入/删除:
- 同源聚合物插入/删除(homopolymer indels)的比例为49.92%,相对较高。同源聚合物区域是指由连续相同碱基组成的区域,例如AAAAA或CCCCC。在这些区域中,测序技术往往更容易出现插入或删除错误。因此,同源聚合物插入/删除的比例较高可能是测序技术本身的局限性所致。
结论与建议:
- 尽管插入和删除事件的总数较低,但同源聚合物插入/删除的比例较高,这可能会对某些特定区域的分析造成一定影响。例如,在变异检测或基因注释时,需要特别注意这些同源聚合物区域。
- 对于后续分析,建议使用专门针对同源聚合物错误的校正工具或方法,以减少这些错误对结果的影响。此外,在解释数据时,应谨慎对待同源聚合物区域的变异,可能需要额外的验证或实验证据来支持结论。
- 总体而言,虽然存在一些同源聚合物插入/删除的问题,但插入和删除事件的整体比例较低,表明测序数据在大多数方面仍然是高质量的。因此,在分析时应综合考虑各种因素,以获得更准确、可靠的结果。
转录组组装
1.cufflinks进行重建全场转录本序列
代码如下(示例):
cufflinks -G /home/lumino/TEST1/genomic.gtf -b /home/lumino/TEST1/GCA_000001735.2_TAIR10.1_genomic.fa -u -p 8 /home/lumino/TEST1/STARoutput3/STAR_result/SRR3418005-filtered-STAR.sorted.bam -o /home/lumino/TEST1/cufflinksoutput
-G /home/lumino/TEST1/genomic.gtf:指定了参考基因组的GTF文件,用于提供基因的注释信息。-b /home/lumino/TEST1/GCA_000001735.2_TAIR10.1_genomic.fa:指定了参考基因组的FASTA文件,用于提供基因组序列信息。-u:表示进行无方向性的转录本组装,即不考虑转录本的方向性信息。-p 8:指定了使用的线程数为8,以加快转录本组装的速度。/home/lumino/TEST1/STARoutput3/STAR_result/SRR3418005-filtered-STAR.sorted.bam:指定了输入的BAM文件,包含了对应样本的比对结果。-o /home/lumino/TEST1/cufflinksoutput:指定了输出结果的文件夹路径,转录本组装的结果将保存在该文件夹中。- 完成后有4个输出文件:
genes.fpkm_tracking:这个文件记录了每个基因的表达水平,以FPKM(每百万个碱基对的片段数)为单位。每行包含一个基因的信息,包括基因ID、基因名称、FPKM值等。isoforms.fpkm_tracking:这个文件记录了每个转录本(isoform)的表达水平,以FPKM为单位。每行包含一个转录本的信息,包括转录本ID、基因ID、FPKM值等。skipped.gtf:这个文件记录了在转录本组装过程中被跳过的转录本的注释信息。这些转录本可能由于低表达或其他原因被排除在外。transcripts.gtf:这个文件记录了转录本组装的结果,包含了每个转录本的注释信息,如转录本ID、基因ID、外显子的边界等。
2.转录本注释文件的比较
为了将cufflinks输出的注释文件与现有模型进行比较,命令如下:
cuffcompare -r /home/lumino/TEST1/genomic.gtf /home/lumino/TEST1/cufflinksoutput/transcripts.gtf
-r /home/lumino/TEST1/genomic.gtf:指定参考的基因组注释文件路径,这里是/home/lumino/TEST1/genomic.gtf。/home/lumino/TEST1/cufflinksoutput/transcripts.gtf:指定待比较的转录组注释文件路径,这里是/home/lumino/TEST1/cufflinksoutput/transcripts.gtf。
本文转载自: https://blog.csdn.net/weixin_43927366/article/details/136549657
版权归原作者 Luminosite Eternelle 所有, 如有侵权,请联系我们删除。
版权归原作者 Luminosite Eternelle 所有, 如有侵权,请联系我们删除。