
文章目录
前言
FASTA和FASTQ文件,其实还是文本文件,用于存储序列信息。平时为了存储方便,节省空间,所以变成了GZ的压缩文件(二进制文件,不能直接用
head
、
less
等命令直接查看)。之前没有好好记笔记,现在补上😑
FASTA
FASTA是一种非常简单的生物序列储存格式,包括核酸序列(DNA/RNA)或者组成蛋白质的氨基酸序列(Amino Acid sequence,简称AA序列)。主要由交替重复的两部分(行)组成:
- 以 “ > ” 为开始的行,主要储存的是序列的描述信息;
- 序列部分,中间前后都可以含有空格
例子:血红蛋白α的核酸和蛋白质序列
# 核酸序列
>gi|13650073|gb|AF349571.1| Homo sapiens hemoglobin alpha-1 globin chain (HBA1) mRNA, complete cds
CCCACAGACTCAGAGAGAACCCACCATGGTGCTGTCTCCTGACGACAAGACCAACGTCAAGGCCGCCTGG
GGTAAGGTCGGCGCGCACGCTGGCGAGTATGGTGCGGAGGCCCTGGAGAGGATGTTCCTGTCCTTCCCCA
CCACCAAGACCTACTTCCCGCACTTCGACCTGAGCCACGGCTCTGCCCAGGTTAAGGGCCACGGCAAGAA
GGTGGCCGACGCGCTGACCAACGCCGTGGCGCACGTGGACGACATGCCCAACGCGCTGTCCGCCCTGAGC
GACCTGCACGCGCACAAGCTTCGGGTGGACCCGGTCAACTTCAAGCTCCTAAGCCACTGCCTGCTGGTGA
CCCTGGCCGCCCACCTCCCCGCCGAGTTCACCCCTGCGGTGCACGCCTCCCTGGACAAGTTCCTGGCTTC
TGTGAGCACCGTGCTGACCTCCAAATACCGTTAAGCTGGAGCCTCGGTGGCCATGCTTCTTGCCCCTTTG
G
# 蛋白序列
>sp|P69905|HBA_HUMAN Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1
MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHG
KKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTP
AVHASLDKFLASVSTVLTSKYR
FASTQ
FASTQ文件其实就是FASTA文件的扩展,把测序的质量信息也存储在文件中。在这种格式中,序列和质量分数都表示为单个ASCII字符。主要由四个包含不同信息的交替重复部分(行)组成:
- 第一行,以@起始的,包含序列信息等可选描述,如测序机器编号等
- 第二行,序列字符
- 第三行,“+”,没错,只有个加号
- 第四行,表征第二行序列的测序质量信息编码(ASCII值),必须包含与第二行序列相同数量的字符。表征参考错误率,可以简单理解为对应位置碱基的质量值,越大则测序质量越好。不同的测序仪,会有不同的标准。
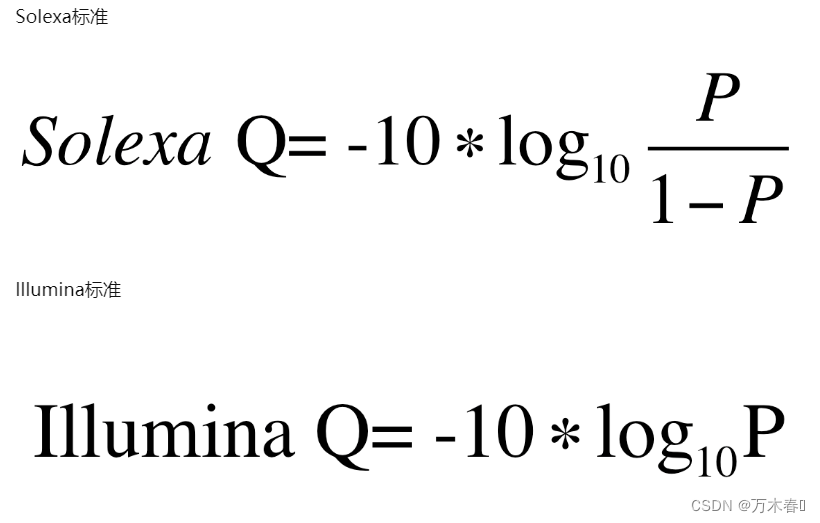
计算方式的细节,参见:https://zhuanlan.zhihu.com/p/20714540
FASTA FASTQ 对比
见图:
FASTQ 转为 FASTA
了解了二者的格式,格式的转换就相对容易,方式的选择就会多种多样。
使用基本的命令:sed、paste、awk
sed
可以用来选择性的输出所需的行,所以,只要在Fastq文件的每4行中,选择打印第1、2行,就可以完成FASTQ到FASTA的转换:
sed-n'1~4s/^@/>/p;2~4p' INFILE.fastq > OUTFILE.fasta
如果不太好理解,也可以配合
paste
命令,将FASTQ的每四行排列为以制表符分隔的一行,然后使用
sed + tr
命令转换:
cat INFILE.fastq |paste - - - - |cut-f1, 2|sed's/@/>/'g |tr-s"/t""/n"> OUTFILE.fasta
有了转换思路,尝试用更全面的
awk
命令将FASTQ转换为FASTA(与其说是命令,我更倾向
awk
是一种语言):
cat infile.fq |awk'{if(NR%4==1) {printf(">%s\n",substr($0,2));} else if(NR%4==2) print;}'> file.fa
简单解释一下,NR代表记录数,循环当中可以简单的理解为 “ 第几行 ” ,或者说是 “ 行号 ” 。在END块内使用,就是文件总记录数(差不多就是总行数)。那么,NR%4就是行号除以4的余数,
head -n 8 tmp.fq | awk '{printf("Line:%s remainder: %s\ncontext: %s\n", NR, NR%4, $0)}'
:分别显示行号(Line),行号除以4的余数(remainder)和该行的内容(context)
Line:1 remainder: 1
context: @A00234:1023:HLGC5DSX3:1:1101:1253:1000 1:N:0:ATCGTACT
Line:2 remainder: 2
context: NGATTGATCGATGAGTAAACAAAGTGAGCAACAGACAGAAAAGGGGCTGTCTCTTATACACATCTCCGAGCCCACGAGACATCGTACTATCTCGTATGCCGTCTTCTGCTTGAAAAATGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG
Line:3 remainder: 3
context: +
Line:4 remainder: 0
context: #FFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFF,FFFFFFFFFFF,FFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFF:FFFFF,FFFFF,,F,FF:F,FF:F,,,,:,,,:::F,:FFFF:FFFFFFFFFFFFFFFFFFFFF
Line:5 remainder: 1
context: @A00234:1023:HLGC5DSX3:1:1101:1542:1000 1:N:0:ATCGTACT
Line:6 remainder: 2
context: NTTTGATTCCGTACTTTGCCTATTCGGTATTCCGAGAGGCTGGTCGGATTGGGTTACAGCCTTGTCTGTATAGGAGCTGTCTCTTATACACATCTCCGAGCCCACGAGACATCGTACTATCTCGTATGCCCTCTTCTGCTTGAAAAATGG
Line:7 remainder: 3
context: +
Line:8 remainder: 0
context: #FFFFFFFFF:FFFFFFFF,FFFFFF:FFFFFFFFFFFFFFFFFFF,FF:FFFFFFF:FFF:FFFFFFFFFFFFFFFF:FFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFF::FFFFFFF,:,F,:FF,,,,,,F,F,FF,,,,
可以看到当余数为1或者2的时候,就是转换需要的,所以此时使用判断语句,当余数等于 1 或者 2 的时候,打印需要的内容。这里还使用了
awk
内置函数
substr
,语法:
substr(input-string, location, length)
,为了去除@符号,所以就从序列的第二位开始打印。
使用现有工具:Bioawk、Seqtk
Bioawk & Seqtk
来自李恒大佬开发的工具,使用方式:
bioawk -c fastx '{print ">"$name"\n"$seq}' input.fastq > output.fasta
seqtk seq-a input.fastq > output.fasta
可以使用压缩文件 (.gz) 作为输入
EMBOSS:seqret
使用方式:
seqret -sequence reads.fastq -outseq reads.fasta
FASTX-toolkit
fastq_to_fasta
函数,适合超大数据:
fastq_to_fasta -h
usage: fastq_to_fasta [-h][-r][-n][-v][-z][-i INFILE][-o OUTFILE]# Remember to use -Q33 for illumina reads!
version 0.0.6
[-h]= This helpful help screen.
[-r]= Rename sequence identifiers to numbers.
[-n]= keep sequences with unknown (N) nucleotides.
Default is to discard such sequences.
[-v]= Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z]= Compress output with GZIP.
[-i INFILE]= FASTA/Q input file. default is STDIN.
[-o OUTFILE]= FASTA output file. default is STDOUT.
FASTA 到 FASTQ
了解前面,也知道,实现这种转换的关键是质量信息,但是,咱没有,所有这时候就需要模拟一个,已经有一些工具帮咱实现了类似功能,比如python工具
pyfastaq
、
bioconvert
等,代码也很简单,比如:
# pyfastaq 默认质量40
fastaq to_fake_qual in.fasta - | fastaq fasta_to_fastq in.fasta - out.fastq
# bioconvert 使用虚拟质量 "I", 并且第一行信息不加其他的东西
bioconvert fasta2fastq in.fa tout.fq
闲聊
重要的是了解格式,掌握之后,格式之间的转换就会得心应手,至于用什么工具,什么语言写的工具,看自己喜欢喽,当然,对于生物信息学,Linux系统、命令行工具暂时还是绕不过去的,必须要掌握。
至于其他语言,只要能达成目的!!
版权归原作者 万木春❀ 所有, 如有侵权,请联系我们删除。