
1. 写在前面
作为现在最火的
scRNAseq
分析包,
Seurat
当之无愧。😘
本期开始我们介绍一下
Seurat
包的用法,先从
基础质控
和
过滤
开始吧。🥳
2.用到的包
rm(list = ls())
library(Seurat)
library(tidyverse)
library(SingleR)
library(celldex)
library(RColorBrewer)
library(SingleCellExperiment)
3. 示例数据
3.1 读取10X文件
这里我们提供一个转成
gene symbols
的可读文件,如果大家拿到的是
Ensemble ID
,可以用之前介绍的方法进行转换。
adj.matrix <- Read10X("./soupX_pbmc10k_filt")

3.2 创建Seurat对象
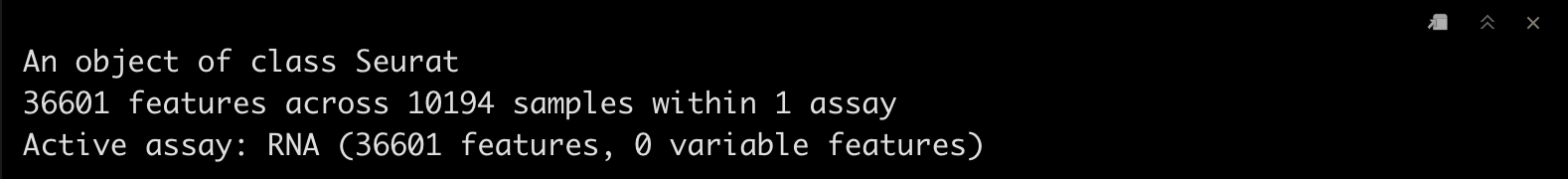
srat <- CreateSeuratObject(adj.matrix,project = "pbmc10k")
srat
3.3 查看Seurat对象
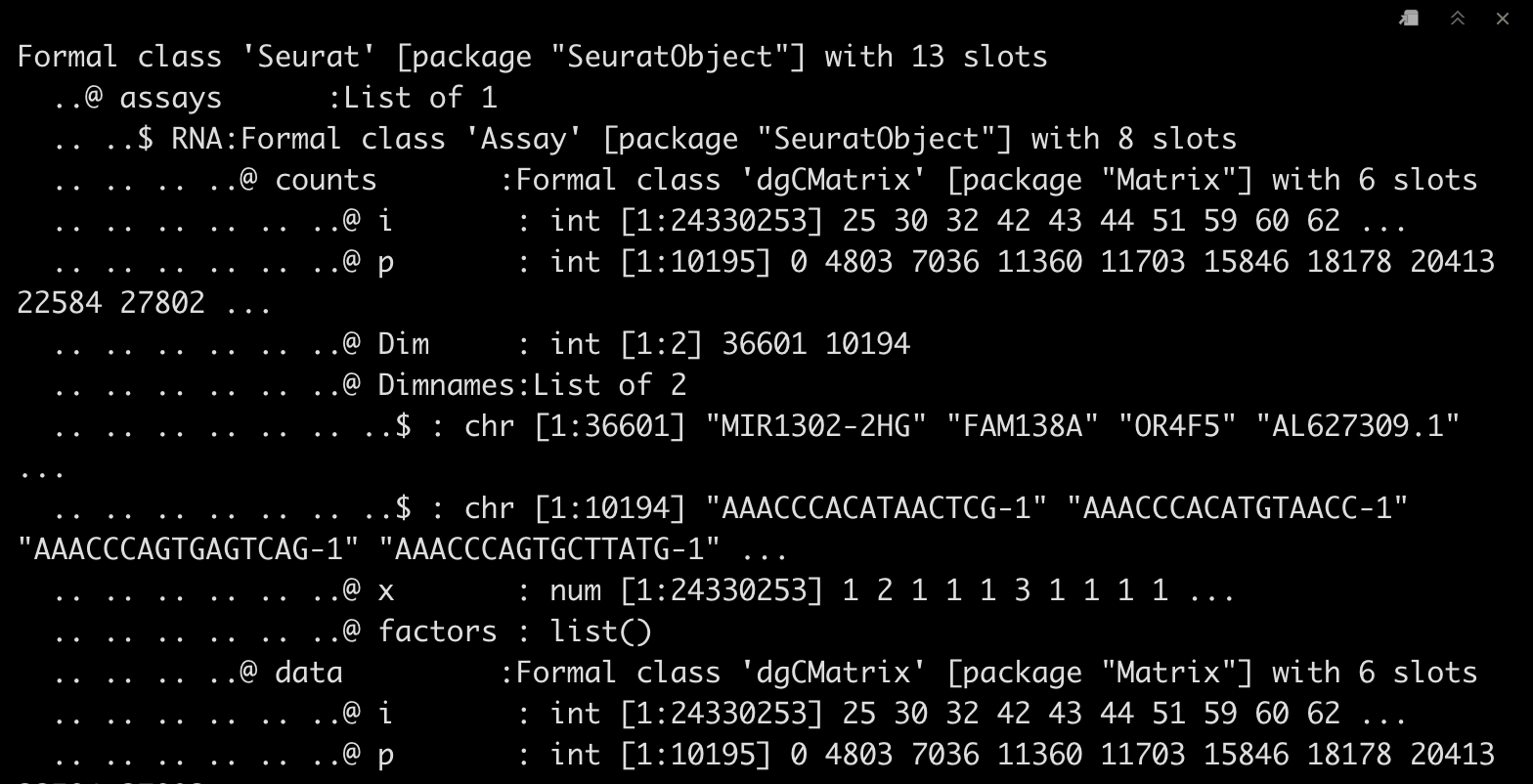
str(srat)
4. 提取meta.data
这里我们提取一下
meta.data
,顺便查看一下表头,主要是三列: 👇
dataset ID;UMI/cell(nCount_RNA);detected genes/cell(nFeature_RNA)。
meta <- [email protected]
head(meta)

5.添加信息
5.1 添加线粒体基因信息
不知道大家还记得线粒体基因吗???🤒
在
scRNA-seq
中,线粒体基因高表达往往代表细胞状态不佳。🧐
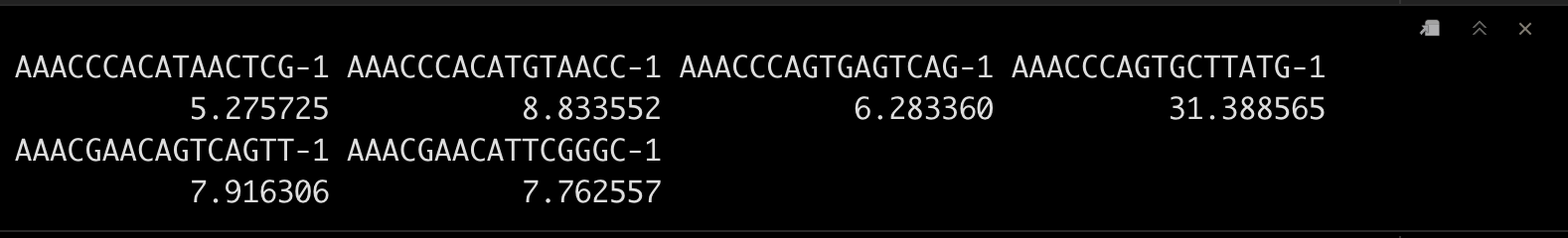
srat[["percent.mt"]] <- PercentageFeatureSet(srat, pattern = "^MT-")
head(srat$percent.mt)
5.2 添加核糖体基因信息
这里我们试一下添加
核糖体基因
的信息。👀
srat[["percent.rb"]] <- PercentageFeatureSet(srat, pattern = "^RP[SL]")
head(srat$percent.rb)

6. 去除双细胞
scRNAseq
的理想情况是每个
barcode
下只有一个细胞,但在实际情况中会有两个或多个细胞共用一个
barcode
,我们称之为
doublets
。🫠
识别并去除
doublets
的方法很多,常用的有:👇
Scrublet;doubletCells;cxds;bcds;Hybrid;DoubletDetection;DoubletFinder;Solo;DoubletDecon。
这里推荐大家使用
DoubletFinder
,我们就不进行演示了,以后再做具体介绍。🤗
因为我们事先使用
Scrublet
做过处理了,这里就直接导入准备好的文件吧。
doublets <- read.table("./scrublet_calls.tsv",header = F,row.names = 1)
colnames(doublets) <- c("Doublet_score","Is_doublet")
srat <- AddMetaData(srat,doublets)
head(srat[[]])

7. 可视化
7.1 小提琴图
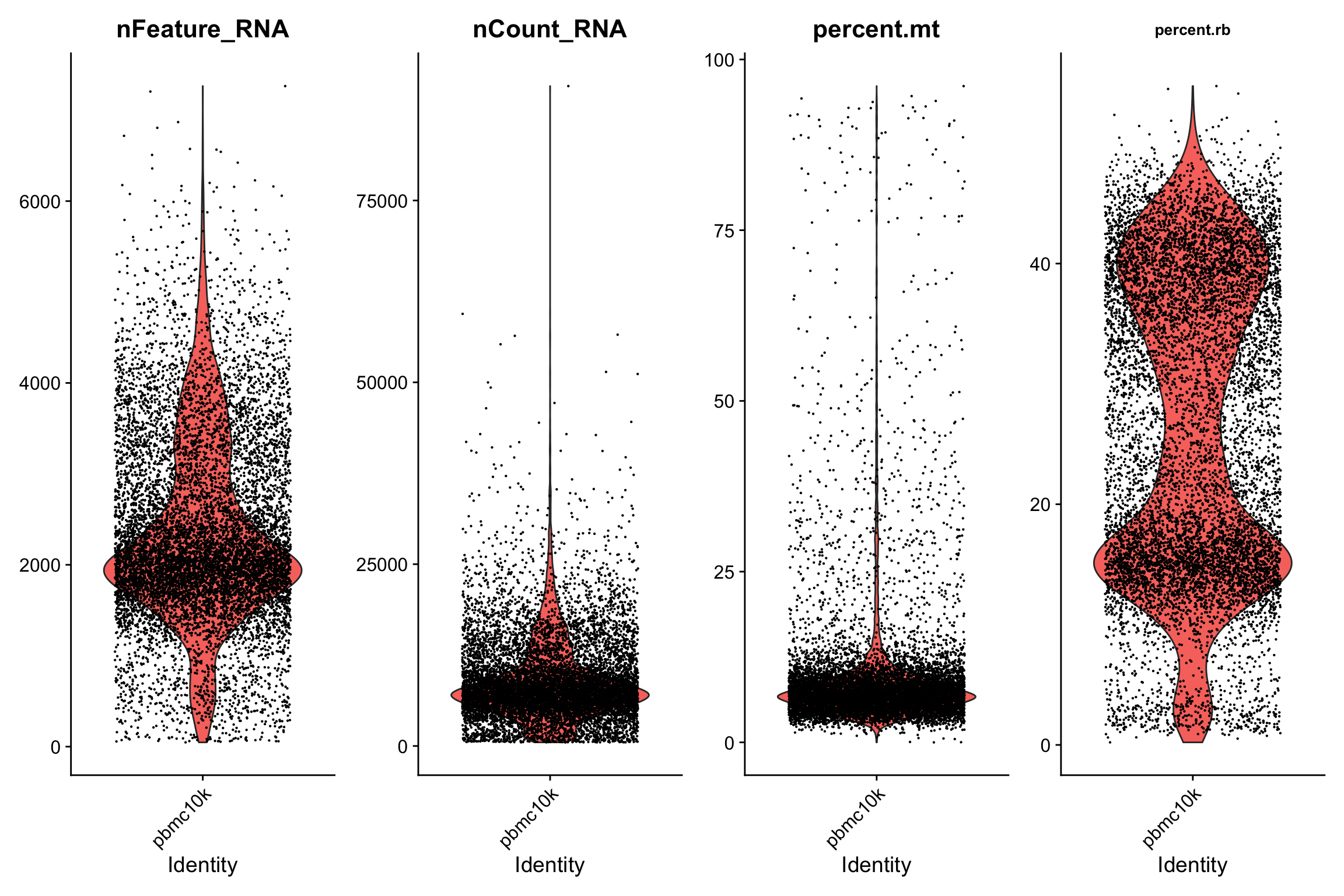
这里我们用
VlnPlot
探索一下特征的分布情况。
VlnPlot(srat,
fill.by = "feature", # "feature", "ident"
features = c("nFeature_RNA","nCount_RNA","percent.mt","percent.rb"),
ncol = 4, pt.size = 0.1) +
theme(plot.title = element_text(size=10))
7.2 散点图
这里利用
散点图
,我们看一下不同变量间的相关性。
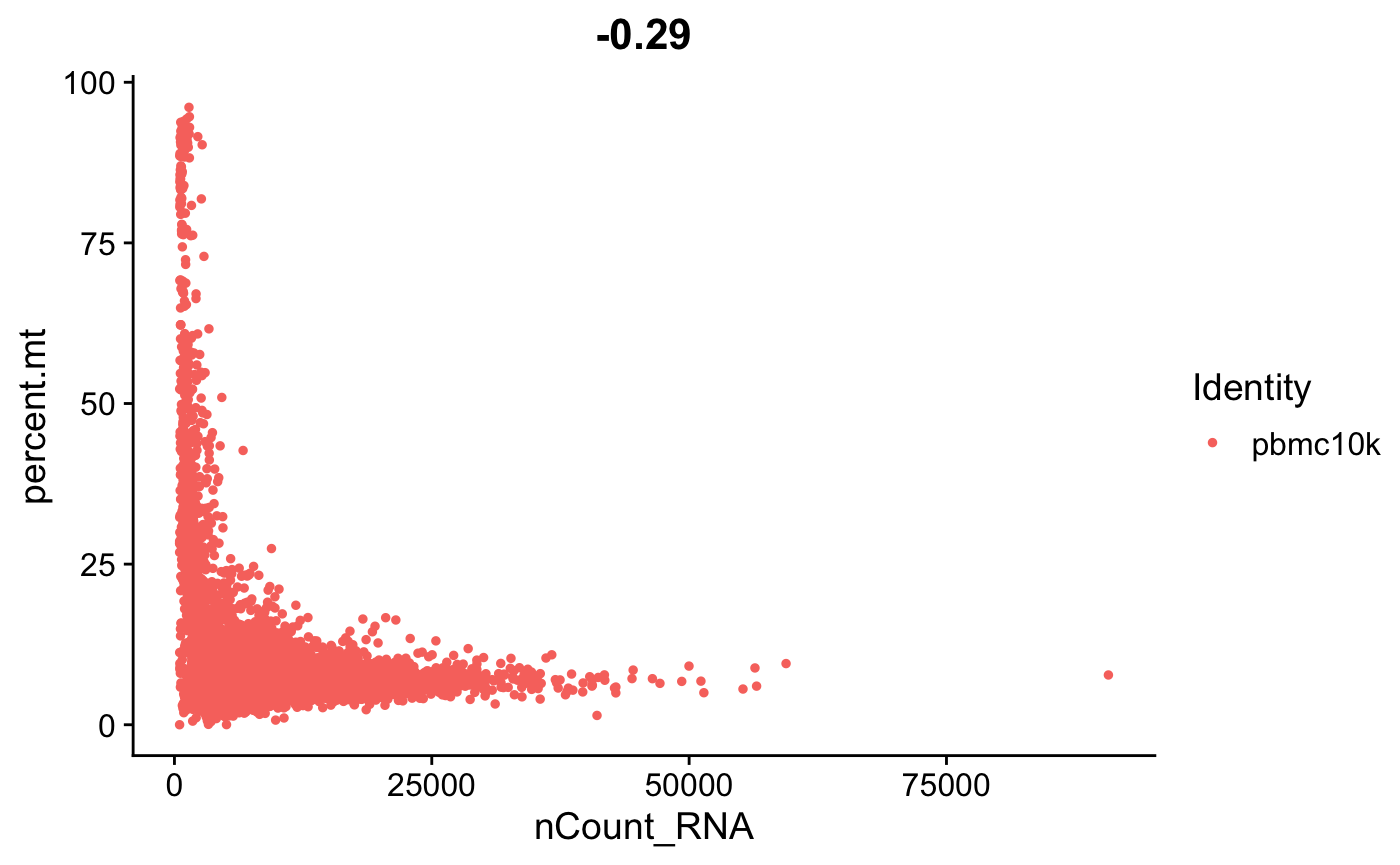
FeatureScatter(srat, feature1 = "nCount_RNA", feature2 = "percent.mt")
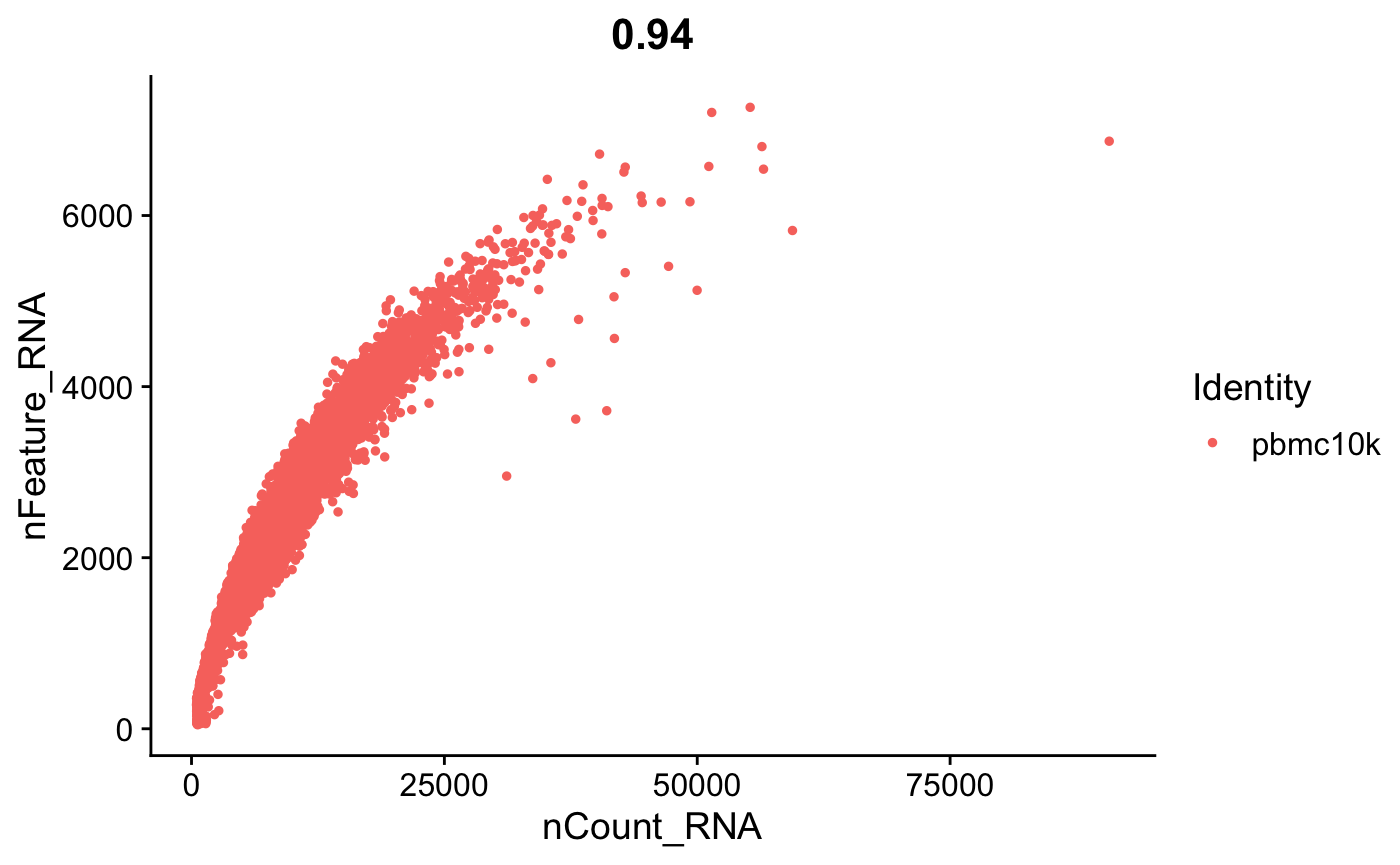
FeatureScatter(srat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
FeatureScatter(srat, feature1 = "nCount_RNA", feature2 = "percent.rb")

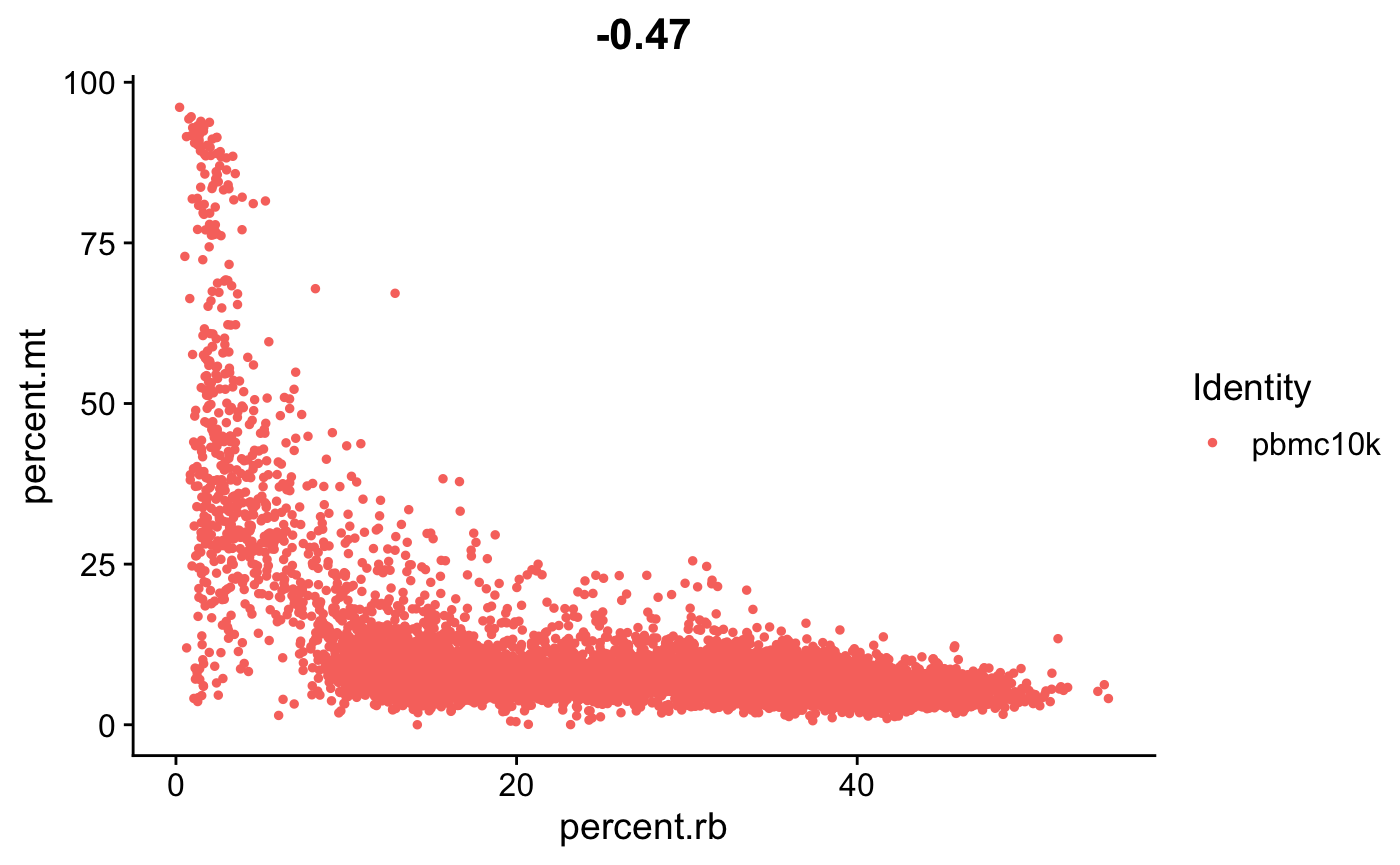
FeatureScatter(srat, feature1 = "percent.rb", feature2 = "percent.mt")
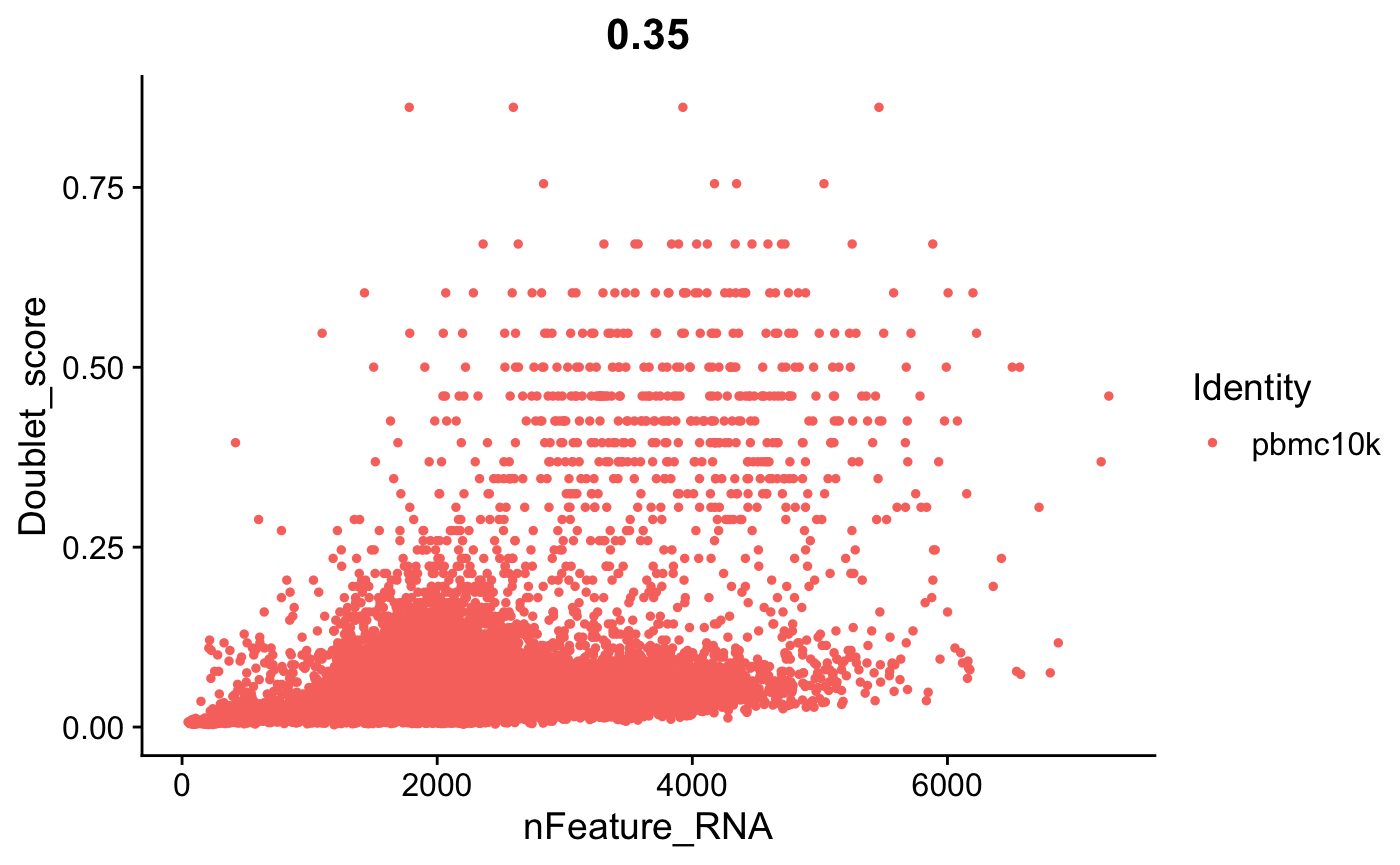
FeatureScatter(srat, feature1 = "nFeature_RNA", feature2 = "Doublet_score")
Note!
- 这里我们可以看到高线粒体基因与低UMI计数相关,可以理解为死细胞。 🫠
- 再看一下核糖体基因与线粒体基因,显著负相关。 😉
doublet和基因表达数之间也有一定的相关性。
8. 添加信息
8.1 过滤
接着我们定义一下过滤条件,将质量差、非单细胞的数据剔除掉。🫵
srat[['QC']] <- ifelse([email protected]$Is_doublet == 'True',
'Doublet','Pass')
srat[['QC']] <- ifelse([email protected]$nFeature_RNA < 500 &
[email protected]$QC == 'Pass',
'Low_nFeature', [email protected]$QC
)
srat[['QC']] <- ifelse([email protected]$nFeature_RNA < 500 &
[email protected]$QC != 'Pass' &
[email protected]$QC != 'Low_nFeature',
paste('Low_nFeature', [email protected]$QC, sep = ','),
[email protected]$QC
)
srat[['QC']] <- ifelse([email protected]$percent.mt > 15 &
[email protected]$QC == 'Pass',
'High_MT',[email protected]$QC
)
srat[['QC']] <- ifelse([email protected]$nFeature_RNA < 500 &
[email protected]$QC != 'Pass' &
[email protected]$QC !='High_MT',
paste('High_MT',[email protected]$QC,sep = ','),
[email protected]$QC
)
table(srat[['QC']])
8.2 可视化
这里我们只将通过过滤条件的数据展示出来,大家可以和过滤前的比较一下。
VlnPlot(subset(srat, subset = QC == 'Pass'),
features = c("nFeature_RNA", "nCount_RNA", "percent.mt","percent.rb"),
ncol = 4, pt.size = 0.1) +
theme(plot.title = element_text(size=10))


最后祝大家早日不卷!~
需要示例数据的小伙伴,在公众号回复
Seurat获取吧!
点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
本文转载自: https://blog.csdn.net/m0_72224305/article/details/127385534
版权归原作者 生信漫卷 所有, 如有侵权,请联系我们删除。
版权归原作者 生信漫卷 所有, 如有侵权,请联系我们删除。