
利用Python读取fasta文件并进行一系列操作(上)
概述
语言:
python3.8
模块:
pysam
collections
可选:
jupyter
整体思路:将fasta格式的基因原始数据处理为方便读写的txt格式并进行操作
步骤:
- 获取自己的
fasta文件(这里我将从NCBI上下载人类的ABO基因参考序列的fasta文件为例)
- 利用
pysam模块的FastaFile函数读取fasta,之后即可获取fasta的基本信息:filename文件名,references染色体编号(因为这里我下载的是ABO基因的fasta,所以不是染色体编号),nreferences染色体数,lengths每条染色体长度(这个非常重要!)
import pysam as sam
# 读取fasta
fasta = sam.FastaFile('ABO_sequence.fasta')# 获取染色体编号
fasta.references
# 获取文件名
fasta.filename
# 获取染色体数
fasta.nreferences
# 获取每条染色体长
fasta.lengths
返回结果如下:
['NG_006669.2']
b'ABO_sequence.fasta'1[42144]
- 通过获取输出的长度,我们知道了这个ABO基因的长度值为42144,我们再利用fetch函数获取ABO基因碱基序列(注意索引是从0开始)
data = fasta.fetch('NG_006669.2',0,42144)
输出结果如下图: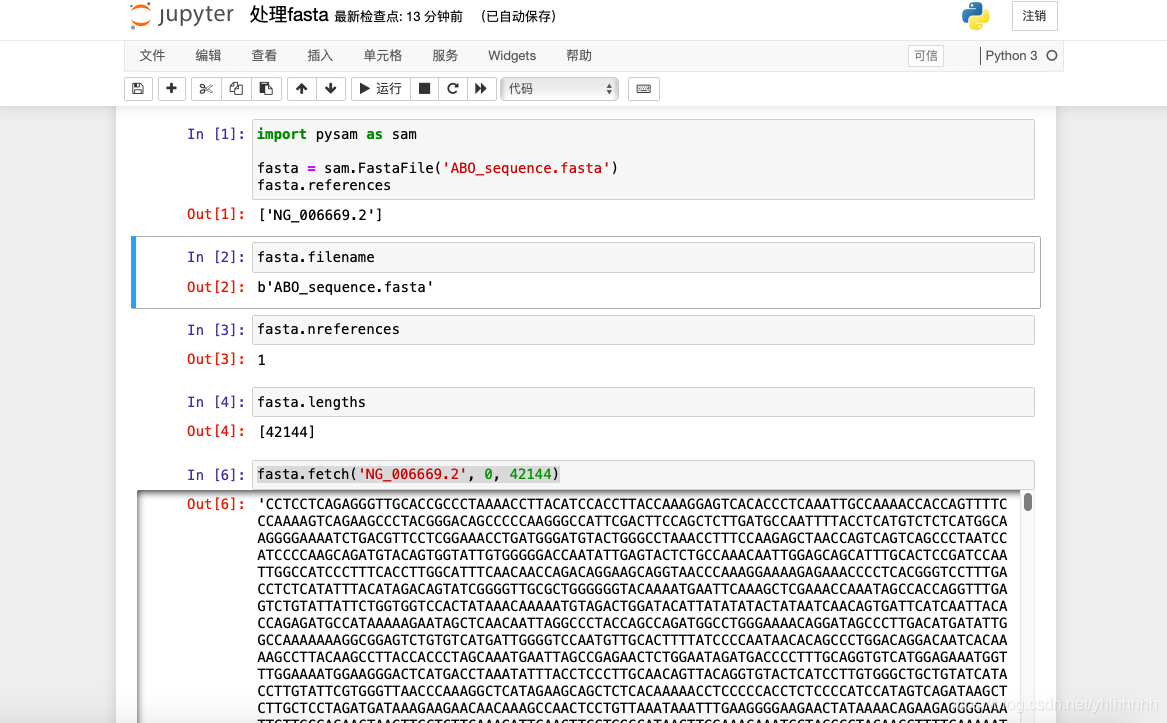
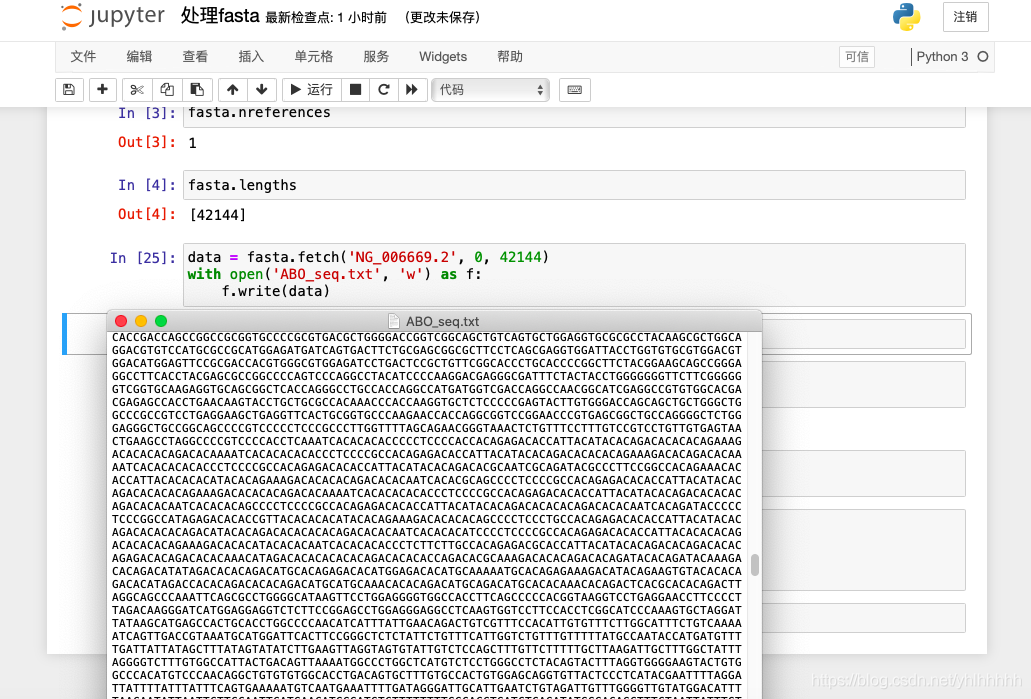
- 我们的最终目的是要输出txt,所以就需要将我们的碱基序列写到txt上
withopen('ABO_seq.txt','w')as f:
f.write(data)
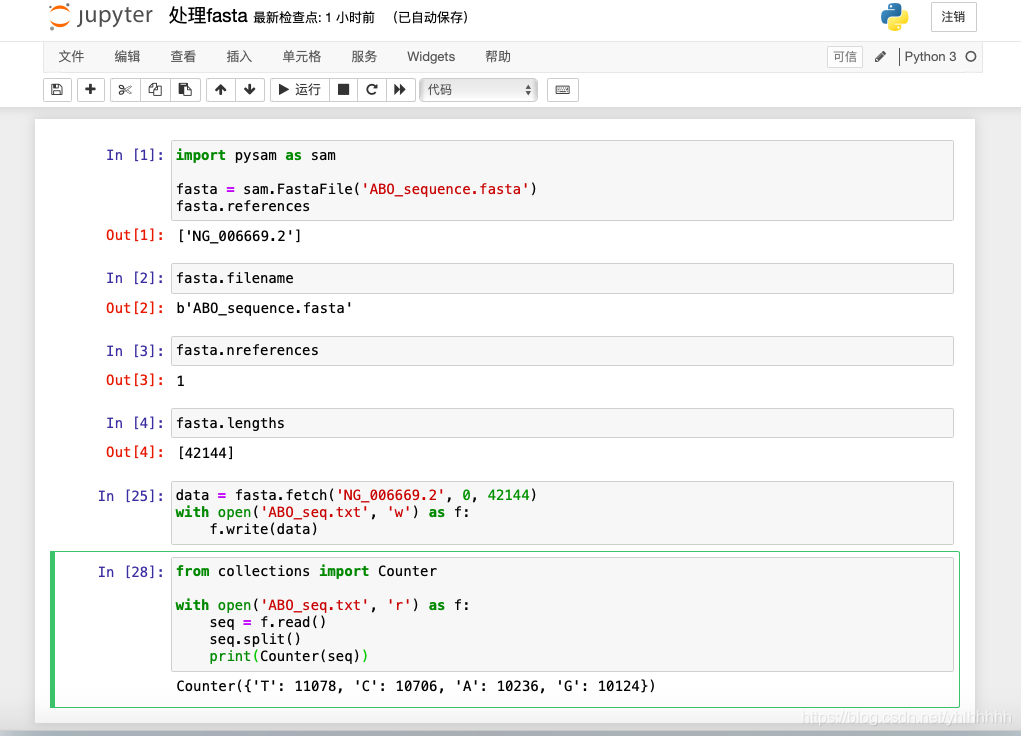
- 我们可以利用我们输出的
txt来做点事情,例如看看这个序列中4种碱基数量,先读取文件,再处理为列表,利用collections模块的Counter函数来输出结果
from collections import Counter
withopen('ABO_seq.txt','r')as f:
seq = f.read()
seq.split()print(Counter(seq))
结果展示:
下面总结一下将fasta输出为txt的代码如下:
import pysam as sam
# 读取fasta
fasta = sam.FastaFile('ABO_sequence.fasta')# 获取每条染色体长
fasta.lengths
# 获取指定的碱基序列
data = fasta.fetch('NG_006669.2',0,42144)# 将序列输出为txt文件withopen('ABO_seq.txt','w')as f:
f.write(data)
更多操作将在下篇解锁!
本文转载自: https://blog.csdn.net/yhlhhhhh/article/details/118034731
版权归原作者 yhlhhhh 所有, 如有侵权,请联系我们删除。
版权归原作者 yhlhhhh 所有, 如有侵权,请联系我们删除。